Circulating Permeability Factors in Idiopathic Nephrotic Syndrome
Article information

Abstract
Nephrotic syndrome (NS) is a common chronic glomerular disease in children characterized by significant proteinuria with resulting hypoalbuminemia, edema, and hyperlipidemia. Renal biopsy findings of diffuse foot processes effacement on electron microscopy and minimal change disease, focal segmental glomerulosclerosis (FSGS), or diffuse mesangial proliferation on light microscopy. It has been speculated that circulating permeability factors would be implicated in the pathogenesis of NS because they have been reportedly detected in the sera of patients and in experimental models of induced proteinuria. Moreover, a substantial portion of the patients with primary FSGS recurrence shortly after transplantation. This report reviews the current knowledge regarding the role of circulating permeability factors in the pathogenesis of proteinuria in NS and suggests future targeted therapeutic approaches for NS.
Introduction
Nephrotic syndrome (NS) is characterized by significant proteinuria (>40 mg/m2/h or a spot urinary protein-to-creatinine ratio of over 2 mg/mg) and hypoalbuminemia (<3.0 g/dL), which in turn causes edema and hyperlipidemia [1-4]. The most common form (90%) of NS in children is primary (idiopathic) and is classified, on the basis of clinical response to steroid therapy, as steroid sensitive or steroid resistant. In terms of pathologic findings, the effacement of diffuse foot processes is common on electron microscopy. Over 80% of childhood patients with NS show minimal change disease (MCD) characterized by normal renal histology on light microscopy, where the remaining 20% show focal segmental glomerulosclerosis (FSGS), diffuse mesangial proliferation, or mesangial proliferative glomerulonephritis [1-4].
Recent advances in podocyte biology have enforced our knowledge of the molecular changes and genetic defects of the glomerular filtration structures, which could explain the mechanism of proteinuria in NS [5-7]. The development of proteinuria in NS is caused by an increased glomerular permeability to plasma proteins, which is accompanied with phenotypical changes of podocytes. The Shalhoub hypothesis [8] suggests that circulating permeability factors could be implicated in the structural and molecular changes of podocytes in idiopathic NS. However, defining a single putative factor remains elusive [1-4]. This paper reviews the current experimental and clinical knowledge of the role of circulating permeability factors in the pathogenesis of proteinuria in idiopathic NS.
Evidence of circulating permeability factors in NS
The pathologic role of circulating permeability factors in podocyte structure and function, leading to proteinuria and NS, is supported by the following clinical and experimental observations [9,10]: (1) a child born from a mother with FSGS and NS spontaneously recovered from proteinuria after maternal transfer of nephrotic proteinuria [11,12]; (2) the perfusion of isolated rat glomeruli with plasma of patients with FSGS induced an increased glomerular permeability to albumin as compared to normal control plasma [13,14]; (3) after kidney transplantation from a donor without the disease, approximately 30% of patients with FSGS developed massive proteinuria within hours to days, followed by FSGS histological lesions [15-17]; (4) patients with recurrent FSGS after kidney transplantation showed complete or partial response to early plasma exchange [18-21]; (5) pre-transplant plasmapheresis or immunotherapy reduced the risk of FSGS recurrence after transplantation [22,23]; (6) the successful transplantation in an end-stage renal disease patient due to diabetic nephropathy of a graft removed from a transplanted FSGS recipient because of intractable recurrence of massive proteinuria and renal insufficiency [24]; (7) the kidneys from a donor with FSGS transplanted into two uremic recipients remained free from proteinuria with normal renal function after one year [25].
Identification
The clinical and experimental evidence of circulating permeability factors suggest that they are involved in the pathogenesis of glomerular hyperpermeability. Maas et al. [26] suggested criteria to define the pathogenic circulating factors in idiopathic MCD and FSGS. In recent decades, research has focused on identifying permeability factors using various models, as will be discussed in the following sections.
Isolation of responsible factors
In 1974, Shalhoub [8] theorized that a disordered clone of T-lymphocytes, present in both MCD and FSGS, secreted a circulating lymphokine “toxic” to the glomerular barrier. The first experimental evidence suggesting the pathophysiological role of lymphokine in idiopathic NS was reported by Lagrue et al. [27] in 1975, showing that the supernatant of lymphocytes from patients with MCD elaborated in vitro by concanavalin A-stimulated T-lymphocytes contained a factor that modified the vascular permeability. The molecular characteristics of the permeability factors have been derived from findings that the active fraction of sera from patients with FSGS precipitates in 70-80% ammonium sulfate solution independent of the immunoglobulin fraction. The putative permeability factors were found to bind to protein A and had a molecular size between 30 and 50 kDa [28].
In vitro experiments
The role of isolated permeability factors could be examined by in vitro endothelial cell or podocyte culture models. Observations of cultured found changes in the configuration of the actin cytoskeleton, cell morphology, and protein expression in responses to permeability factors. Sera from patients with FSGS recurrence were found to disrupt podocyte focal complexes via immunofluorescence [29]. Hemopexin was found to affect the podocyte actin cytoskeleton and reduced endothelial glycocalyx, thereby increasing albumin diffusion [30,31].
Ex vivo experiments
Glomerular permeability could be measured using ex vivo isolated glomeruli developed by Savin et al. [32]. In isolated rat glomeruli, when an isotonic albumin oncotic solution was replaced by a solution with a lower albumin concentration, an increase in glomerular size was observed through swelling when the permeability barrier was intact. It was found that serum from patients with FSGS increased the permeability of glomeruli to albumin [28]. Recently, Desideri et al. [33] improved the ex vivo model and found that incubation with plasma from patients with post-transplant recurrence of NS increased albumin permeability in rat glomeruli compared to remission plasma. The blocking effect of galactose on circulating permeability factors was demonstrated using ex vivo isolated glomeruli [34]. A glomerulus-on-a-chip microdevice is a recently developed ex vivo model, which could replace isolated glomeruli [35].
In vivo experiments
Plasma permeability factors have been found to induce proteinuria in experimental animals. Zimmermann [13] demonstrated that serum from patients with recurrent FSGS after renal transplantation could induce increased albumin excretion when infused in the aorta of intact rats. Koyama et al. [36] found that human T cell hybridomas formed from cells from patients with MCD produced a substance that induced proteinuria and reduced the density of polyethylamine staining of glomerular anionic sites when injected intravenously into normal Wistar rats. Each permeability factor, such as hemopexin [37] and cardiotrophin-like cytokine factor-1 (CLCF-1) [38], also induced proteinuria when injected intravenously into normal rats.
Suggested permeability factors
Although the clinical and experimental data suggest that a circulating permeability factor induces glomerular proteinuria and NS, a single putative factor has yet to be identified. Several candidates are implicated in the development of proteinuria in idiopathic MCD and FSGS [9,10,26,39-41]. The proposed candidates are listed in Table 1. Clinical and experimental data have suggested roles for cytokine interleukin (IL)-13, CD80, anti-CD40 antibody, angiopoietin-like 4 (angptl-4), circulating CLCF-1 (member of the IL-6 family), circulating hemopexin, and soluble urokinase-type plasminogen activator receptor (suPAR), among others, in the development of NS.
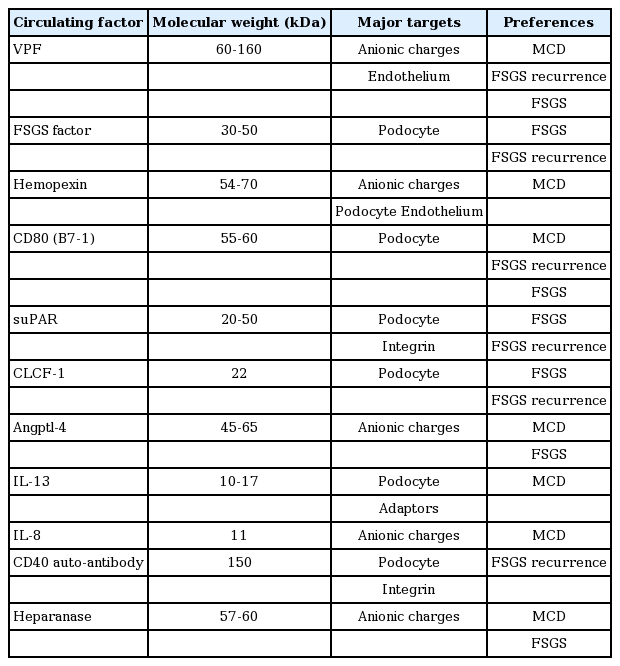
Circulating Permeability Factors Proposed in Idiopathic Nephrotic Syndrome
Vascular permeability factor (VPF)
Lagrue et al. [27] were the first to systematically demonstrate the existence of a factor that could modulate vascular permeability. They defined potential plasma factors on vascular permeability after finding that VPF was produced in vitro after the stimulation of isolated blood T-lymphocytes with concanavalin A. Lymphocyte-derived VPF following concanavalin A stimulation in vitro was found to reduce glomerular polyanions by colloidal iron staining following in vitro incubation of kidney cryostat sections, and was found to cause proteinuria in rats [42,43]. Concanavalin A-derived supernatants of lymphocytes from patients with MCD induced proteinuria in rats in vivo following infusion into the renal artery [44] or tail vein [36,45]. However, lymphocyte-derived VPF has failed to be characterized despite the continuous production of supernatants. Although VPF was first identified as a permeability factor in MCD, the following studies actually showed that VPF was present in most but not all patients with MCD and was not unique to MCD. Moreover, there is no direct evidence that VPF causes proteinuria or that it even has an effect on glomerular permeability [26].
FSGS factor
The FSGS factor was identified from observations wherein the active fraction of sera from patients with recurrent FSGS was found to induce proteinuria in rats [13]. Sera from patients with recurrent FSGS increased glomerular permeability to albumin compared to normal control plasma in the perfusion of isolated rat glomeruli model [14,28], and was found to disrupt podocyte focal complexes [29]. The active fraction of permeability factor precipitates in 70–80% ammonium sulfate solution, inconsistent with immunoglobulin fraction. This putative FSGS factor has a molecular size between 30 and 50 kDa and is bound to protein A [28], which is consistent with a report of proteinuria remission after protein A column ex vivo adsorption [20]. This FSGS factor was partly identified as CLCF-1 by sequential purification and fractionation.
Hemopexin
Hemopexin is a serine protease predominantly produced in the liver. It has been suggested that, in normal conditions, circulating hemopexin is inactive; however, under certain circumstances, hemopexin becomes activated as a serine protease [46]. Activated hemopexin appears to alter the function of both glomerular endothelium and podocytes. In vivo, activated hemopexin injection in rats caused reversible proteinuria with podocyte foot processes fusion [37,46]. In cultured podocytes, hemopexin activated protein kinase B and Ras homolog gene family, member A (RhoA), and induced nephrin-dependent reorganization of the actin cytoskeleton from stress fibers to cytoplasmic aggregates and membrane ruffles [30,44]. In addition, hemopexin reduced the anionic sites in the lamina rara interna and endothelial sialoglycoproteins, thereby increasing albumin diffusion across the monolayers of the glomerular endothelial cells [31,47]. Studies on human MCD have demonstrated a decreased titer of plasma and urine hemopexin and an increased activity of circulating isoform during disease relapse which then disappeared during remission [46]. These changes were ascribed to an altered configuration of hemopexin in relapsing MCD. No conclusions have been drawn on the role of hemopexin in MCD, and its implication is, at the moment, believed to be a secondary effect.
CD80 (B7-1)
Cluster of differentiation 80 (CD80, also called B7-1) is a transmembrane molecule present on the surface of B cells and other antigen-presenting cells. When bound to its ligand, CD28, present in T-lymphocytes leads to lymphocyte activation [48]. CD80 expression in dendritic cells is inhibited by cytotoxic T-lymphocyte-associated-protein 4 (CTLA-4) and IL-10, which are produced by T regulatory (Treg) cells [49,50]. Under normal conditions, CD80 is only transiently expressed after trigger stimuli, and the resulting proteinuria is minimal due to rapid auto-regulatory response of circulating Treg cells or the podocyte itself, leading to the expression of factors such as CTLA-4 to downregulate the podocyte CD80 response [51]. The level of circulating CTLA-4 is low during relapses [52] and there is a predominance of certain CTLA-4 genotypes in idiopathic NS [53,54]. Furthermore, CD80 has been proposed to be used for differential diagnosis between MCD and FSGS [55].
The increased expression of CD80 by trigger stimuli results in actin reorganization, as well as a change in the shape of podocytes, leading to the effacement of the foot processes, and proteinuria [56]. Trigger events for increased CD80 podocyte expression in experimental animals include inflammatory stimuli, such as lipopolysaccharide (LPS) and polyinosinic-polycytidylic acid (Poly:IC), oxidative stress, in the case of PAN and IL-13. LPS-injected mice were found to have an increased podocyte CD80 expression through signaling processes mediated by Toll-like receptors (TLR) and IL-13 with foot processes effacement and proteinuria in the early phase which mimicked MCD [48,57]. In contrast, such findings were not observed in CD80 knockout mice when injected with LPS. Furthermore, severe combined immunodeficient (SCID) mice still developed proteinuria in response to LPS, thus showing that CD80 expression and proteinuria are not dependent on T-lymphocytes [58]. The expression of podocyte CD80 induced by LPS in vitro through binding to TLR3 and TLR4 [58] or TNFα treatment [59] has also been associated with actin derangement and shape changes.
The in vitro incubation of human podocytes with poly-IC, a TLR 3 ligand, increased the podocyte expression of CD80 with the activation of TLR3 and cathepsin L, but decreased the expression of synaptopodin, and resulted in actin reorganization via an NF-κB-dependent pathway, which was partially blocked by dexamethasone [48]. An In vivo poly:IC injection also reduced synaptopodin expression in podocytes with proteinuria and increased glomerular CD80 expression and urinary CD80 in mice [60].
The CD80 levels in podocytes and urine were elevated in MCD patients during the initial onset compared to MCD in remission. CD80 was also detected in the glomeruli of the same patients but were not found in high levels in other NS-causing nephropathies, such as FSGS or membranous nephropathy (MN) [61,62]. These results were confirmed in other populations with MCD [63], thus leading to the conclusion that high CD80 (B7-1) expression in the urine and glomeruli is a urinary biomarker to distinguish between MCNS and FSGS, which could be used to monitor the activity of the disease [55,66]. Other studies documented the upregulation of glomerular CD80 (B7-1) in patients with diabetic nephropathy [64] and/or with post-transplant FSGS recurrence [65]. Sera from MCD patients in relapse, but not in remission, stimulated CD80 expression in cultured podocytes along with diminished nephrin phosphorylation in comparison with podocytes incubated with sera from MCD patients in remission [66]. Therefore, B7-1 may be a biomarker of podocyte injury.
The upregulation of glomerular CD80 (B7-1) in patients with post-transplant FSGS recurrence and active MCD led to the use of abatacept (CTLA-4-Ig), a co-stimulatory inhibitor of B7-1, as a specific therapeutic agent for patients with post-transplant FSGS recurrence and MCD. However, the administration of abatacept resulted in a transient reduction of urinary CD80 levels and resolution of proteinuria in an MCD patient [67] and substantially reduced proteinuria in FSGS [65]. The results from other studies were also discordant and did not confirm the original observation [68]. The positive data on B7-1 is being challenged by recent discordant findings obtained in clinical data. As such, further controlled studies on the roles of B7-1 and abatacept are needed in various clinical and experimental glomerular pathological conditions.
Soluble urokinase-type plasminogen activator receptor (suPAR)
Urokinase plasminogen activator receptor (uPAR), a cell membrane glycosylphosphatidylinositol (GPI)-anchored membrane glycoprotein, has a role in the migration of activated T-lymphocytes, monocytes, and neutrophils to sites of inflammation [69-71]. uPAR consists of three domains (DI, DII, and DIII), which are encoded by separate exon sets of the Plaur gene, and bind to their ligand, urokinase plasminogen activator (uPA) [9,71,72]. Through interactions with transcellular receptors, such as integrins, uPAR mediates cell adhesion to vitronectin and activates cell signaling pathways via a mechanism that includes the lipid-dependent activation of αvβ3 integrin for cell migration, proliferation, and survival [73]. The activation of uPAR is associated with cancer cell invasion and migration [74]. These findings led to predict the role of uPAR in motile podocytes.
Through the cleavage of uPAR from its GPI-anchor, suPAR is released. Further cleavage between the DI and DII/DIII domains of suPAR generates other cleaved suPAR fragments. As suPAR and uPAR are heavily glycosylated proteins, the molecular weight (MW) of uPAR ranges from 35 to 60 kDa, whereas the MW of suPAR ranges from 20 to 50 kDa, depending on the amount of glycosylation and the size of the cleaved proteins [9,26,39]. Notably, these MW ranges are similar to those of the circulating permeability factor proposed by Savin [39].
The suPAR, expressed by several immune cells and endothelial cells, but also by podocytes, can induce foot processes effacement and FSGS in vitro and in vivo and has been shown to be increased in the plasma of FSGS patients compared to patients with other glomerular diseases, inclusive of MCD [55,75]. Podocyte foot processes contain an actin-based cytoskeleton that is bound to the vitronectin molecules of the glomerular basement membrane (GBM) via α3β1 and αvβ3 integrins [71,76]. The activation of vitronectin-dependent αvβ3-integrin induces intracellular signaling processes, leading to the development of podocyte foot processes and the modifications of the actin cytoskeleton [76]. Podocyte uPAR binds to integrin and vitronectin. In podocyte cultures and murine models, uPAR was found to cause vitronectin-dependent αvβ3-integrin activation [77].
Wei et al. [75] found that the serum concentrations of suPAR were significantly elevated in patients with primary FSGS. However, patients with MCD, MN, and preeclampsia did not display a significant elevation of suPAR levels. In addition, suPAR levels correlated with the presence but not with the level of proteinuria. The highest concentrations of suPAR were found in patients with recurrent FSGS after transplantation. Elevated levels of suPAR prior to transplantation appears to increase the risk of recurrence of disease in the transplanted kidney, and preliminary evidence suggests that treatment with plasmapheresis may significantly reduce these levels and induce remission [75]. Wei et al. [78] also defined 3,000 pg/mL as the cut-off value for a high probability of diagnosing FSGS and found that approximately two-thirds of patients with primary FSGS, and to a lesser degree patients with proteinuric kidney disease, were found above this level [78]. In a FSGS CT (70 adults) and Podonet (94 children) cohort study, the authors reported that suPAR levels were elevated in 84.3% (CT cohort) or 55.3% (Podonet cohort) of patients with primary FSGS, and there was an inverse correlation of suPAR levels with estimated glomerular filtration rate (eGFR) [78]. However, in the two studies, the serum suPAR data were not corrected for eGFR. Li et al. [79] also confirmed in their cohort (109 primary FSGS, 20 MCD, 22 MN, and 96 healthy controls) that suPAR levels were elevated in about half of their patients with FSGS, and could therefore discriminate between FSGS and other proteinuric kidney diseases. In addition, the suPAR levels indicated the steroid-responsiveness of FSGS [79]. However, serum suPAR levels did not differ between primary and secondary FSGS [80]. The initial findings of suPAR as a potential causal factor in primary FSGS was a major breakthrough, and many researchers have subsequently tested the serum suPAR levels in human adult [80-84] and pediatric [85-87] cohorts with conflicting results after controlling suPAR values for eGFR.
The urinary suPAR level of patients with primary FSGS were also significantly higher compared to those with MCD, MN, secondary FSGS, or normal subjects [88], and were positively correlated with 24-hour urinary protein excretion in primary FSGS. During the follow-up, the urinary suPAR level of patients with complete remission decreased significantly. After incubation of human podocytes with urine obtained from patients with primary FSGS at presentation, the AP5 signal was induced and then reduced by a blocking antibody specific to uPAR [88].
The pathological role of suPAR on the development of FSGS has been demonstrated in both in vivo and in vitro experimental models. Wei et al. [75] established three different mouse models and confirmed that suPAR caused proteinuria and FSGS in mice by binding to the podocyte β3 integrin. In Plaur −/− mice, infusion with over 20 μg of recombinant mouse suPAR protein induced podocyte deposition of suPAR with an increase in β3 integrin activity and proteinuria within 24 hours. Furthermore, wild-type mice with transplanted kidneys from Plaur −/− mice modeling endogenous suPAR release developed proteinuria after being challenged with LPS, indicating that circulating suPAR may be able to activate β3 integrin independently of uPAR. In the third model, wild-type mice with plasmid transfer (sPlaur WT, suPARI-II producing plasmid) into the skin showed a rise of serum suPAR and FSGS-like lesions with proteinuria. However, mice that received a gene transfer for the defective β3-integrin binding suPAR mutant, sPlaur E134A, did not become albuminuric, suggesting that binding of suPAR to β3 integrin is important in suPARinduced renal injury. Subsequently, the mouse uPAR isoform 2 was found to induce FSGS with proteinuria using suPAR2 transgenic mice, and β3 integrin was found to play a critical role in the development of renal dysfunction in suPAR2-transgenic mice in three different integrin β3 deficiency models [89]. Alfano et al. [90] also showed that high dose (20 μg) of suPAR (full-length recombinant murine suPAR) induced proteinuria in Plaur −/− mice and suPAR decreased nephrin expression in podocytes via the suppression of Wilms tumor-1 (WT-1) transcription factor In vivo and in vitro . Interestingly, only the full-length suPAR molecule interacted with β3-integrin and caused podocyte damage, however, cleaved suPAR molecules were not able to activate β3-integrin. On the other hand, Delville et al. [91] revealed that injecting suPAR (recombinant human suPAR) did not induce proteinuria in wild-type mice unless injected in combination with other podocyte-damaging agents, such as anti-CD40 antibodies. In contrast, Spinale et al. [83] did not detect proteinuria in wild-type mice 24 hours after the administration of recombinant suPAR (recombinant mouse suPAR-Fc), despite high suPAR levels. Furthermore, the ectopic expression of the full-length suPAR (DI-DIII) molecule from the liver did not induce proteinuria for 44 days despite the elevated suPAR levels. Similarly, Cathelin et al. [92] found that neither the short-term nor prolonged administration of suPAR (recombinant mouse suPAR-Fc and monomeric mouse uPAR produced in S2-cells) could induce proteinuria in wild-type mice [59].
Many questions remain regarding the potential pathogenic role of plasma and serum suPAR in primary FSGS. However, from the evidence presented above, suPAR appears to be a biomarker for reduced renal function. Further studies will be needed to clarify the role of suPAR in podocyte damage in primary FSGS patients to provide the novel treatment strategies for FSGS patients [9,40,71,93-95].
Cardiotrophin-like cytokine factor-1 (CLCF-1)
CLCF-1, a member of the IL-6 family of cytokines with a predicted MW of 22 kD, is expressed by several tissues and is known to activate B cells [96]. CLCF-1 is secreted and forms heterodimers with either cytokine receptor-like factor 1 (CRLF1) or soluble ciliaric neurotrophic receptor alpha (sCNTFRα), resulting in a composite cytokine [97]. CLCF-1 was isolated by galactose affinity chromatography and found in the active fraction of plasma from patients with recurrent FSGS, at levels up to 100 times higher than the controls [36]. CLCF-1 decreased nephrin expression in glomeruli and podocytes [96]. The incubation of murine podocytes with CLCF-1 disrupted the actin cytoskeleton in time- and concentration-dependent manners and led to a motile podocyte phenotype [96]. Similar to unfractionated FSGS serum, CLCF-1 isolated by galactose affinity chromatography induced albuminuria in mice and increased albumin permeability in isolated rat glomeruli through the activation of the JAK/STAT pathway, however, these experiments did not exclude the involvement of other galactose-binding permeability factors [96]. This effect can be reversed by incubation with a monoclonal antibody against CLCF1 or with JAK/STAT inhibitors, suggesting that antibodies targeting CLCF-1 or its receptors and JAK2/ STAT3 inhibitors may be novel therapeutic agents to treat FSGS [97]. Recombinant human CLCF1 increased the albumin permeability in isolated rat glomeruli, which was inhibited by a heterodimer composed of CLCF1 and co-secreted molecule cytokine receptor-like factor 1 [97].
Angiopoietin like-4 (angptl-4)
Angptl-4 is a 45 to 65 kDa glycoprotein and acute phase reactant expressed in adipose tissue, liver, skeletal muscle, and the heart, and exists in two isoforms, with a high (pI>8) and a neutral (pI=7) isoelectric point, depending on the sialic acid content [98,99]. A hyposialylated form secreted from podocytes can induce proteinuria in animal models, through an effect on either the GBM or endothelial cells, and by reducing the anionic charges of the glomerular filtration barrier. In contrast, a neutral pI sialylated form is released into the circulation from other tissues and can reduce proteinuria by binding αvβ5 integrins on the glomerular endothelium; it also induces hypertriglyceridemia via the inhibition of lipoprotein lipase. Rats overexpressing angptl-4 outside the kidney have high circulating levels of this factor but do not develop proteinuria, however, mice with podocyte overexpression of angptl-4 have normal circulating levels of the protein and do develop proteinuria [100]. Glomerular angptl-4 expression and urinary angptl-4 levels also increased in diabetic rats [101]. In healthy individuals, angptl-4 is minimally expressed in the glomeruli, but is upregulated in the serum and urine of MCD patients [102]. Initial studies found that high circulating angptl-4 levels and glomerular expression coincided in human MCD and was related to proteinuria. However, subsequent studies have reported conflicting results.
The serum levels of angptl-4 were also elevated in patients with FSGS and MN compared with the controls, in addition to active MCD [98]. In a recent study, Cara-Fuentes et al. [103] found that there was no correlation between serum angptl-4 levels and proteinuria in MCD, FSGS, and MN patients during relapse. Furthermore, the serum angptl-4 levels were decreased in MCD patients during relapse compared with the same patients in remission and the controls. In contrast with a previous study, no or minimal angptl-4 staining by immunofluorescence was observed in kidney tissue from MCD patients in relapse. They also found that urinary angptl-4 excretion in relapsed patients correlated with proteinuria in the FSGS and MN groups, however, this positive correlation was not found in MCD patients during relapse. Increased urinary angptl-4 excretion may be the result rather than the cause of proteinuria. In addition, no increased angptl-4 expression was observed in human podocytes cultured with sera from MCD patients in relapse compared with those exposed to sera from MCD patients in remission. Therefore, the clinical significance of angptl-4 in MCD remains to be determined given the contrasting findings between the relapse and remission groups. Nonetheless, angptl-4 remains a strong candidate for explaining MCD both in human and in experimental models. How sialylated angptl-4 could be used to treat NS is yet to be determined, however, off-target effects, such as the inhibition of lipoprotein lipase leading to hypertriglyceridemia in NS, could be exacerbated [98,100]. Previous studies have demonstrated that the modification of soluble angptl-4 sialylation or of key amino acids can be successfully used to treat proteinuria [104,105].
Interleukin-13 (IL-13)
IL-13 is a type of T cell-derived cytokine [106]. IL-13 has been reported to be increased during MCD relapse, and the expression of the IL-13 gene is elevated both in CD4 and CD8 lymphocytes in children with steroid-sensitive NS during recurrences [107,108]. This increase is associated with higher levels of IL-13 in the cytoplasm of T-cells [108] and the downregulation of IL-8 and IL-12 pro-inflammatory cytokines in monocytes [109]. In addition, Wistar rats overexpressing IL-13 developed clinical NS and showed glomerular lesions identical to those observed in MCN, including an 80% effacement of podocyte foot processes via electron microscopy, the downregulation of nephrin, podocin and dystroglycan, and an increase in podocyte expression of CD80 [57]. We recently found pathological molecular changes in podocyte proteins by IL-13. We previously reported that IL-13 increased podocyte permeability through the modulation of ZO-1 [110], modulated the changes of cytoskeletal and adaptor proteins [111], and increased podocyte apoptosis [112], resulting in proteinuria and podocyte injury.
However, the supposed role of circulating IL-13 in proteinuria in MCD patients is questionable because the elevated serum IL-13 levels were not found in MCD patients during relapse [66] and have been reported in patients with NS during remission compared with the controls [113]. In addition, although serum IL-13 is increased in patients with asthma and with atopic dermatitis, only a few present associated proteinuria [114,115]. Further experimental and clinical research on the patho-mechanism of IL-13 on the development of proteinuria and podocyte injury is needed.
Interleukin-8 (IL-8)
A pathogenic role for IL-8, a chemokine, in glomerular permeability was suggested after serum levels of IL-8 were found to be increased in MCD in relapse compared with those observed during remission and in the controls [116,117]. The urinary IL-8 level was 2.9-fold higher in NS patients in relapse compared to patients in remission, and levels had a positive correlation with individual proteinuria values. Urinary IL-8 was also significantly higher in relapsed steroidsensitive NS patients than in steroid-sensitive NS patients in remission [118]. Rats infused with IL-8 that reached a serum IL-8 concentration similar to that observed in MCD patients developed mild proteinuria, and the administration of anti-IL-8-neutralizing antibody prevented proteinuria, suggesting a causal relationship between IL-8 and proteinuria [119]. In this model, IL-8 increased GBM glycosaminoglycan catabolism and reduced anionic charges at the GBM [120]. However, rat podocytes incubated with high concentrations of human IL-8 did not show any alteration in permeability, heparan sulfate (HS) proteoglycan gene expression, or HS synthesis [117]. Unfortunately, further studies on the role of IL-8 for the development of proteinuria in NS have not been reported.
CD40
CD40 belongs to the TNF receptor superfamily and is expressed preferentially in B cells, monocytes/macrophages, dendritic cells, platelets, as well as non-immune cells, such as endothelial and smooth muscle cells, and is involved in the adaptive immune response [121]. The binding of CD40 to CD40 ligand expressed in T cells and platelets, that is, the CD40/CD40L complex, mediates pro-inflammatory events [122]. The CD40 ligand activates endothelium and leads to increased expression of chemokines, metalloproteases, uPA, and suPAR [123]. CD40 is also constitutively expressed in podocytes, where activation by CD40L promotes the redistribution of nephrin and increases permeability to albumin in isolated glomeruli [123-5]. CD40 was detected in the glomeruli and serum anti-CD40 antibodies were detected in recurrent FSGS patients, with levels which correlated with the post-transplant recurrence of the disease. As such, anti-CD40 antibodies may help to identify patients at high risk of post-transplant outcome [123]. Even though anti-CD40 antibodies from recurrent FSGS sera did not detect recombinant human CD40, purified anti-CD40 antibodies from recurrent FSGS sera were found to disrupt the podocyte actin cytoskeleton in vitro . This finding points to a post-translational modification of the CD40 molecule in vivo, which is necessary for detection with anti-CD40 antibodies [123]. The authors also suggested that anti-CD40 antibodies play a pathogenetic role in the development of recurrent FSGS, potentially via its interaction with suPAR, facilitating or prolonging suPAR-mediated integrin activation, thus potentiating enhanced podocyte motility and disruption of the slit diaphragm [123]. Injection with the CD40/anti-CD40 complex was found to induce proteinuria in mice, while infusion with anti-CD40L antibodies reduced proteinuria and the progression of adriamycin nephrosis [126,127]. The size of IgG anti-CD40 antibodies is approximately 150 kDa [95], which contradicts previous findings where the active fraction of FSGS sera was found to be 30-50 kDa [28,40]. Anti-CD40 blocking antibodies (ASKP1240 or lucatumumab) are already commercially available and could become potential treatment options to be tested in clinical trials [41].
Heparanase
Heparanase, an endo-β (1-4)-D-glucuronidase that cleaves the glycosidic bond within the HS chain via hydrolysis, is involved in various biologic processes. Glomerular heparanase degrades HS polysaccharide side chains in GBM and thereby induces proteinuria via the loss of anionic charges, alterations in the glomerular cell-GBM interactions due to loss of HS, and the release of HS-bound factors, such as growth factors, cytokines, and chemokines, and bioactive HS fragments in glomeruli [128].
Heparanase has been found to play an important role of in several proteinuric diseases. A disturbance in heparanase expression was detected in the urine and glomeruli of proteinuric patients with MCD, FSGS, MN, and diabetic nephropathy [128-132]. In steroid-sensitive NS in children, Holt et al. [129] found increased heparanase expression in the kidney glomeruli, increased heparanase concentration in the urine, and decreased heparanase concentration in the serum. A discrepancy between serum and urinary heparanase was associated with the accumulation of heparanase deposits along the vascular walls, the presence of inhibitors in the serum, or a non-proportional increase in heparanase versus changes in proteinuria. The importance of heparanase has also been demonstrated in the several forms of experimental glomerular diseases. Increased heparanase expression and/or decreased HS was found in the early stages of experimental anti-GBM antibody disease [133], passive Heymann nephritis as an experimental MN model [134-136], and puromycin aminonucleoside or adriamycin nephropathy [137,138]. The expression of heparanase was found to remain stable over the course of these disorders.
However, the role of heparanase in the development of proteinuria has recently been challenged. A strong reduction in glycosaminoglycan-associated anionic sites in GBM without renal changes casts doubt on the primary role of HS in charge-selective filtration, suggesting that an increase in heparanase expression and a decrease in HS in GBM in glomerular diseases is a consequence rather than a cause of proteinuria [128,139]. Indeed, proteinuria-lowering interventions, such as the inhibition of the renin-angiotensin-aldosterone system or reactive oxygen species scavengers, which are known to be unrelated to heparanase, also induced a regression of heparanase expression [138]. Adriamycin-injected wild-type mice developed severe albuminuria, while ADR-injected mice with transgenic heparanase overexpression exhibited only mild proteinuria and preserved podocyte phenotypes. Therefore, heparanase may have a nephroprotective role in adriamycin-induced NS, most likely via a mechanism independent of HS degradation [140]. No further studies on the role of heparanase in the development of proteinuria have been carried out.
Conclusions
The biologic advances in our understanding of the pathogenesis of podocytopathies have facilitated the identification of various podocyte molecules and several candidate mediators that could play a role in FSGS or MCD. The pathogenic sequences of the permeability factors involved in the development of proteinuria and NS are presented in Fig. 1. Despite variations in the severity of correlation and the type of NS, permeability factors in MCD are VPF, hemopexin, CD80, angptl-4, heparanase, IL-13, and IL-8. On the other hand, suPAR, CLCF-1, and CD40 autoantibodies are permeability factors that would be more applicable in FSGS. However, none of these molecules have been identified as the unique primary cause of either MCD or FSGS [6,9,26,40,95]. Currently, new drugs are being developed that target podocyte molecules and pathogenic mediators involved in the pathogenesis of MCD/FSGS and recurrent FSGS.

The pathophysiology of proteinuria and nephrotic syndrome.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This report was supported by the Clinical Research Fund of Chungbuk National University Hospital and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2013R1A1A4A03006207 and 2016R1D1A1B 03931888).