Acute Tubular Necrosis associated with the Ketogenic Diet in a Child with Intractable Epilepsy
Article information

Abstract
The ketogenic diet (KD) has been used as an effective antiepileptic therapy for intractable childhood epilepsy. However, various adverse effects have been reported with use of the KD. We report a case of a child who developed acute tubular necrosis subsequent to therapy with KD. A 5-year-old girl had myoclonic epilepsy with developmental delay. She was under the treatment with antiepileptic drugs since the age of 3 months and on the KD during the past 18 months. Proteinuria persisted intermittently with the initiation of the KD and subsequently increased in the past 2 months. She was admitted with intermittent mild fever, vomiting, and lethargy for the past 3–4 weeks. At the time of admission, she presented with hypertriglyceridemia, heavy proteinuria, renal Fanconi syndrome, and acute kidney injury. Renal sonography showed a marked increase in the size and parenchymal echogenicity of both kidneys. A renal biopsy revealed acute tubular necrosis accompanied by early interstitial fibrosis. After the withdrawal of the KD and supportive therapy, without changing other anticonvulsants and their dosages, improvement of renal function was observed. Proteinuria had disappeared after 1 month and kidney size returned to normal after 8 months. It is hypothesized that the KD can induce and/or aggravate the renal tubulointerstitial injury in some patients who are under the treatment with anticonvulsants.
Introduction
The ketogenic diet (KD) has emerged over the past decade as an important therapeutic option for intractable childhood epilepsy [1,2]. Since the active application of KD after the mid-1990s, many kinds of early and late-onset complications have been reported [3]. There have been several documents about kidney stones developing in children on the KD, and other renal complications including Fanconi renal tubular acidosis has been shown in clinical and experimental studies [3-6]. In the present study, we describe a rare case of persistent proteinuria, renal Fanconi syndrome, and acute tubular necrosis (ATN) associated with the KD in a 5-year-old girl with intractable epilepsy.
Case report
A 5-year old girl was admitted with intermittent mild fever, vomiting, and lethargy for the past 3–4 weeks. She was born by vaginal delivery at 38 weeks of gestation, and her birth weight was 3,160 g. She had been treated for myoclonic seizures since the age of 3 months. Because of intractable seizures, she had been treated with the formula-based KD during the past 18 months. Antiepileptic therapy also included topiramate 150 mg/day, oxcarbazepine 450 mg/day, clobazam 10 mg/day, divalproex sodium 375 mg/day, rufinamide 600 mg/day, quetiapine 12.5 mg/day, L-carnitine 660 mg/day, and pyridoxine 100 mg/day. She had no other systemic diseases except for the developmental delay and mental retardation. Metabolic and mitochondrial disorders were excluded throughout the metabolic and genetic tests. Several measurements of plasma lactate and pyruvate levels revealed no abnormal findings. Mild proteinuria persisted intermittently with the initiation of the KD; however, renal function and serum albumin were normal. Hypertriglycedemia had been observed irregularly (15 months ago, triglycerides (TG) 207 mg/dL; 9 months ago, TG 316 mg/dL). Two months back, she developed hypokalemia, metabolic acidosis, and proteinuria and at that time, laboratory findings revealed serum protein 6.4 g/dL, albumin 3.9 g/dL, blood urea nitrogen 15.9 mg/dL, creatinine 0.31 mg/dL, sodium 143 mEq/L, potassium 2.9 mEq/L, chloride 112 mEq/L, total CO2 12.8 mmol/L, and a dipstick urine protein 2+. At the time of visit to our clinic, she was lethargic and dehydrated and had lost 10% of her body weight. Vital signs were body temperature 36.8℃, pulse rate 100/min, respiration rate 22/min, and blood pressure 98/50 mmHg. Laboratory findings revealed white blood cell count 10,400/μL, hemoglobin 8.5 g/dL, and platelets 466×103/μL. Other parameters were, serum glucose 83 mg/dL, protein 5.1 g/dL, albumin 2.8 g/dL, AST 64 IU/L, ALT 40 IU/L, lactic dehydrogenase 664 IU/L, blood urea nitrogen 33.7 mg/dL, creatinine 1.08 mg/dL, sodium 144 mEq/L, potassium 1.3 mEq/L, chloride 110 mEq/L, total CO2 10.7 mmol/L, ionized calcium 4.7 mg/dL, phosphorus 2.0 mg/dL, cholesterol 143 mg/dL, TG 405 mg/dL, amylase 48 IU/L, and lipase 337 IU/L. A urinalysis showed a specific gravity of 1.018, pH 6.0, glycosuria (1+), proteinuria (3+), and 3-plus ketones. There were no nitrites, pyuria, and hematuria. The random urine protein to creatinine ratio was 5,690 mg/g, suggesting heavy proteinuria. Urinary β2-microglobulin was also high (β2-microglobulin to creatinine ratio 103,875 μg/g, normal <300 μg/g). The serologic tests for C3, C4, CH50, C1q, anti-neutrophil antibody, anti-dsDNA antibody, anti-glomerular basement membrane antibody, and anti-neutrophil cytoplasmic antibody showed all negative results. An increase in plasma ketone bodies was observed (acetoacetic acid 2.41 mmol/L; normal range, 0.05–0.15 mmol/L; β-hydroxybutyric acid, 3.85 mmol/L; normal range, 0.05–0.3 mmol/L), but no enhancement in the serum valproate level was observed (19.93 μg/mL; normal range, 50–100 μg/mL). Renal sonography and computed tomography showed cortical echogenicity and marked increase in the size of both the kidneys (left kidney 9.48 cm, right kidney 8.8 cm) (Fig. 1A and B). On her 6th day of hospitalization, improvement in initial dehydration findings was observed. However, hypokalemia, hypophosphatemia with hyperphosphaturia, metabolic acidosis, heavy proteinuria, and glycosuria persisted irrespective of supportive therapy and the laboratory parameters were: blood urea nitrogen 16.9 mg/dL, creatinine 0.62 mg/dL, sodium 145 mEq/L, potassium 2.4 mEq/L, chloride 124 mEq/L, phosphorus 2.0 mg/dL, fractional excretion of phosphate 37.3%, HCO3– 11.4 mmol/L, random urine protein to creatinine ratio 5,720 mg/g, and 1+ glycosuria. We diagnosed her with renal Fanconi syndrome with possible metabolic complications such as hypertriglyceridemia. The KD was discontinued on the 7th day of hospitalization and supportive therapy was continued. A renal biopsy showed shortening or loss of the brush borders and flattening of the proximal tubular cells, resembling distal tubules (Fig. 2A). Interstitial edema with early fibrosis and a few infiltrates of lymphocytes were observed. Homogeneous or granular casts were observed in the dilated tubules and in the Bowman’s spaces, which were periodic acid Schiff stain-positive and bluish on Masson’s-trichrome stain (Fig. 2B). Immune reactants were entirely negative in the glomeruli. Tubular casts were stained with IgA (3+), kappa (3+) and lambda (3+) light chains and fibrinogen (4+). Electron microscopy showed a few swollen mitochondria in the cytoplasm of detached proximal tubular cells. The tubular cells had hydrophic and vacuolar degeneration of the cytoplasm (Fig. 2C). These findings were typical of ATN, accompanied with early interstitial fibrosis, which was most probably drug induced. After the withdrawal of the KD and massive supportive therapy, and without any changes in other anticonvulsants and their dosages, improvement in her general condition and renal tubular function was observed. She was discharged on the 19th day of hospitalization with decreased proteinuria (random urine protein to creatinine ratio 1,390 mg/g). After 1 month, the proteinuria had disappeared (random urine protein to creatinine ratio 170 mg/g) regardless of continuation of the same medication including valproate. The sizes of both the kidneys returned to normal after 8 months (left kidney 7.65 cm, right kidney 6.94 cm) (Fig. 1C).
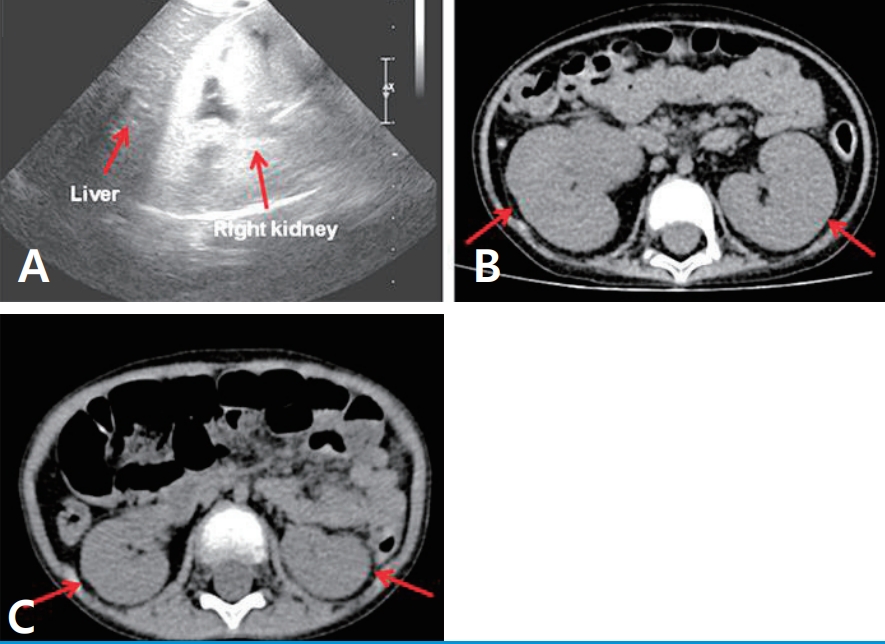
(A) Renal cortical echogenicity appears to be distinctly greater than liver parenchymal echogenicity on renal sonography (B) Both the kidneys are strikingly enlarged on abdominal computed tomography (arrows). (C) Renal size became normal 6 months later (arrows).
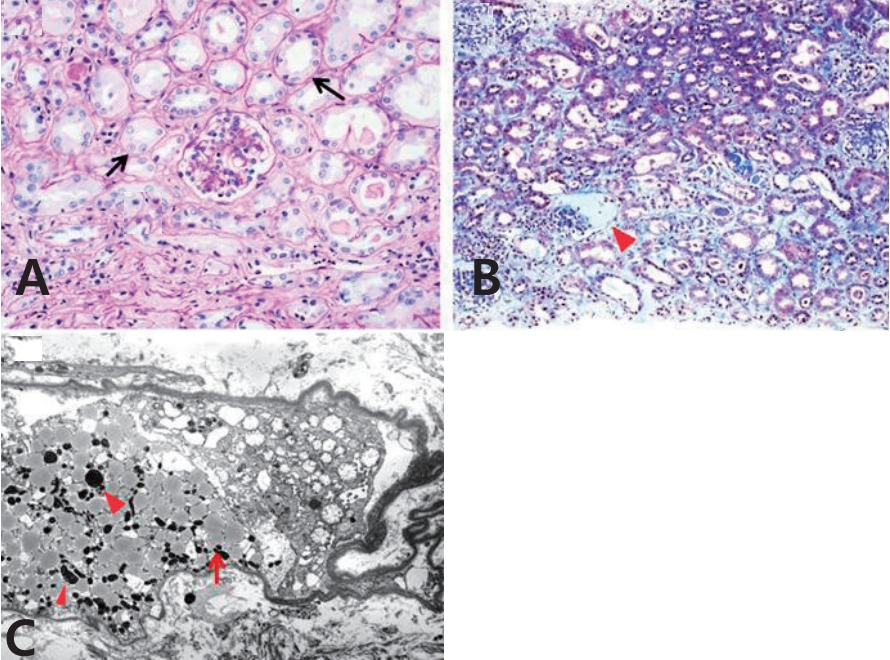
(A) The proximal tubules reveal shortening of the brush border and flattening (arrows) (hematoxylin and eosin stain, ×400). (B) Hyaline casts (arrowhead), interstitial edema, and early fibrosis can be visualized (Masson-trichrome stain, ×200). (C) Electron micrograph shows some swollen mitochondria (arrows) as well as many lipid droplets (arrowheads) (×3,000).
Discussion
The patient reported in the present case had been on the KD during the past 18 months for intractable epilepsy. She had no other underlying systemic disorders. Although she had taken multiple anticonvulsants for more than 4 years, proteinuria developed with the initiation of the KD and aggravated with the development of metabolic acidosis and hypokalemia during the past 2 months. At the time of admission, she was dehydrated and hypertriglyceridemic, and had developed ATN and renal Fanconi syndrome. Her general condition and laboratory findings finally improved after withdrawal of the KD.
The KD, also known as the long-chain TG diet, is a high-fat, adequate-protein, and low-carbohydrate diet, which contains a 4:1 ratio by weight of lipid to non-lipid [1]. It was first introduced in 1921 and since then, it has been used as a potent antiepileptic treatment for intractable childhood epilepsy [1,3]. If there is scant carbohydrate in the diet, the liver converts fat into fatty acids and ketone bodies. An elevated level of ketone bodies in the blood can reduce the frequency of epileptic seizures [7]. Since the active application of KD in therapeutics, many kinds of complications have been reported in numerous studies, some of which had not been described previously [3]. Long-term adverse effects of the KD include growth retardation, gastrointestinal symptoms, carnitine deficiency, kidney stones, and elevated level of lipids. Additional serious adverse effects have been reported in a small number of patients which include cardiomyopathy, Fanconi renal tubular acidosis, and pancreatitis [1,3,8].
Overall, excess fatty acids accompanied by the accumulation of TG results in chronic cellular injury, which is now termed as lipotoxicity [9]. TGs are toxic due to the presence of nonesterified fatty acids (NEFAs) and increase in their products due to the failure of esterification of the TGs [10]. NEFA-induced mitochondrial dysfunction plays an important role in the impairment of pancreatic β-cells and skeletal myocytes. In the kidney, the NEFA-induced mitochondrial injury is the primary mechanism for energetic failure of proximal tubules during hypoxia and/or reoxygenation. Filtered NEFA bound to albumin can aggravate the chronic tubular injury and inflammatory phenotype that develops during proteinuric states. Acute toxicity of NEFA and chronic lipotoxicity can also promote AKI and chronic kidney disease [9]. It is believed that prolonged KD in our patient might have induced excessive TG accumulation along with NEFA-induced tubular injury, which could have contributed to the renal pathology and AKI. A renal biopsy in our case showed the characteristic proximal tubular cell injury accompanied by the findings of drug-induced ATN. Although we excluded the possibility of underlying mitochondrial disorders through several tests, one may argue that a patient with mitochondrial problems who is placed on the KD can develop significant acidosis, which can intermittently worsen with higher energy requirements.
Even under such circumstances, we should not rule out that the possibility that KD can aggravate the mitochondrial injury and contribute to renal tubular injury. Numerous therapeutic agents can negatively affect the kidney, causing tubulointerstitial, glomerular or vascular disease. The tubular injury persists as long as the drug is present, and resolves with withdrawal of the drug [11]. Our patient had taken multiple anticonvulsants for the past 4 years; however, laboratory findings including heavy proteinuria improved after discontinuation of the KD even with the constant use of same anticonvulsants at respective dosages. Enlarged kidney size also returned to normal after stopping the KD. Although valproic acid has been associated with renal Fanconi syndrome [12,13], it may not be a direct cause in our case; as the blood levels were normal and it was taken persistently even after stopping the KD. It is hypothesized that an infectious condition might have contributed to initial dehydration findings with AKI in the present case. Despite acute illness such as viral infection being one of the original causes of her clinical course, the findings from serology tests and renal biopsy did not suggest the infection as a main cause of ATN. Most importantly, the findings of renal tubular injury improved with the discontinuation of the KD. This clinical course suggests that anticonvulsants and/or acute infectious illness may not be a direct cause for ATN, although there could be a potential negative synergistic effect associated with the diet.
Acute pancreatitis has also been reported as a life-threatening complication of the KD [3]. Although serum amylase level was normal in our patient, serum lipase activity was elevated. Hypertriglyceridemia competitively interferes with the amylase assay and can lead to falsely low results [14]. Compared with serum amylase, serum lipase activity remains increased for a longer duration, thereby imparting greater sensitivity to patients with a delayed presentation [15]. As we did not check the serial activities of serum amylase and lipase, we could not confirm the diagnosis of acute pancreatitis in our clinical setting.
Collectively, it is proposed that the KD can increase renal lipotoxicity. The increase in TGs with prolonged use of the KD may affect the development of proximal tubular dysfunction and subsequent tubulointerstitial renal injury. Although most of the adverse effects caused by KD improve with conservative therapy, we experienced conditions of serious illness, which demanded discontinuation of the KD. To the best of our knowledge, this is the first report of biopsy-proven ATN associated with the KD. Early discovery of serious complications and active intervention seem to be critical to prevent fatal outcomes related to the KD.
Notes
Patient consent
The study was approved by the institutional review board (IRB No. 2018GR0312) and the consent was waived due to the nature of the retrospective study.
Disclosure
Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interest
No potential conflict of interest relevant to this article was reported.