Nephronophthisis
Article information
Abstract
NPHP is the most common monogenic cause of CKD in children or adolescents. Extra-renal symptoms often accompany, therefore examination of retina, hearing, and skeleton is necessary in patients with CKD with insidious onset. Genes involved in NPHP-RC are mostly related in primary cilia. While genetic diagnosis is necessary for definitive diagnosis, there is no curative treatment.
Nephronophthisis (NPHP), meaning ‘disappearance of nephrons’, is the most common monogenic cause of chronic renal failure (CRF) in children or adolescents [1-4]. For instance, among 160 end-stage renal disease (ESRD) patients of our center, one fourth had clinical diagnosis of NPHP.
Symptoms
NPHP typically has insidious onset, therefore when the diagnosis is made, most of the patients have advanced chronic kidney disease (CKD) with decreased renal function. Advanced CRF is accompanied by anemia and growth retardation, thus most of NPHP patients present with general weakness, pallor, and poor growth. History taking commonly reveals recent onset polyuria/nocturia and polydipsia, which is considered as resulting from decreased renal concentrating ability. Urinalysis is often normal [5], and the blood pressure is usually not high in early stage. Contrast to autosomal recessive or autosomal dominant polycystic kidney disease (ARPKD or ADPKD), kidney size of NPHP is usually normal or relatively small [6]. On ultrasonography, corticomedullary differentiation of the kidney is lost. Case 1 describes a typical case of juvenile NPHP.
1. Case 1
A 14 years old boy presented with both lower leg pain. He had suffered from general weakness, pallor, nocturia for some time. Laboratory test revealed anemia and azotemia. USG showed small kidneys with increased echogenicity and impaired perfusion (Fig. 1). He denied history of UTI, and his VCUG was normal.
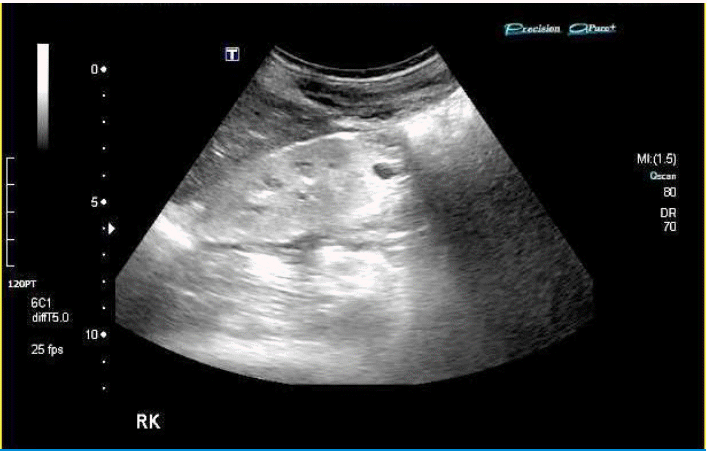
Ultrasonography of the kidney in Case 1 showing typical finding of NPHP. Kidney size of NPHP is usually normal or relatively small, and corticomedullary differentiation of the kidney is lost.
NPHP is often accompanied by extra-renal symptoms, such as retinitis pigmentosa (RP) or Leber’s congenital amaurosis (LCA) in Senior-Løken syndrome (SLSN), hypo-/ a-plasia of cerebellar vermis in Joubert syndrome (JBTS), hepatic fibrosis in Meckel Gruber syndrome (MKS), and COACH (cerebellar vermis hypoplasia/ aplasia, oligophrenia, congenital ataxia, ocular coloboma, and hepatic fibrosis) syndrome with multiple problem [7], listing a few. Because most of the causative genes in these disorders encode proteins that play a role in the cilium [8-10], a collective term NPHP-related ciliopathy (NPHP-RC) is used to describe this group of diseases. Case 2 describes a case of COACH syndrome.
Case 2
A one year old girl presented with developmental delay and apraxia. Imaging studies revealed molar tooth sign' in brain stem (Fig. 2), multiple cysts in the kidneys and hepatic fibrosis. She lost her kidney function when she was 11 years old.

Brain MRI of Case 2 showing ‘molar tooth’ sign.
Diagnosis
Diagnostic criteria of NPHP are as follows [11].
Clinical diagnostic criteria of NPHP
Insidious onset of CRF the first thirty years, without identifiable cause.
Patients often presents with <30 years, idiopathic
Anemia, growth retardation
Polyuria, nocturia, polydipsia from Decreased concentrating ability of the kidneys.
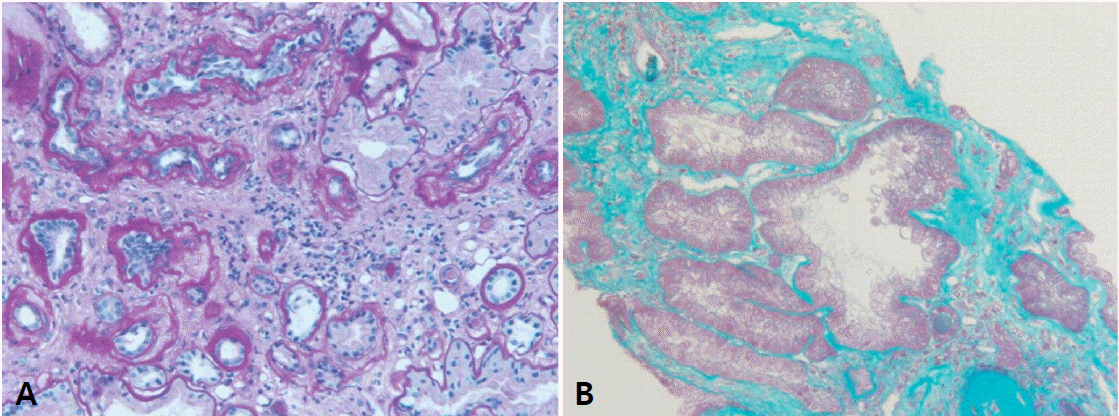
Histological findings are non-specific showing chronic tubulointerstitial nephropathy (Fig. 3), therefore pathologic diagnosis is not mandatory for NPHP. Furthermore, since most of the patients present at their advanced stage, renal biopsy can be risky of bleeding complication. On electron microscopy, tubular basement membrane change is , similar to glomerular basement membrane change in Alport syndrome [12].
Since the clinical features of patients with NPHP-RC are rather non-specific, a genetic diagnosis is required for a definitive diagnosis of NPHP-RC. In addition, multiple syndromes of NPHP-RC share their phenotype in various degrees, which requires clarification of genetic aberration for appropriate diagnosis [10]. Inheritance pattern of NPHP-RC is commonly autosomal recessive (AR), and currently more than 20 causative genes of NPHP are known, with ‘NPHP’ as part of their names [13-15]. Table 1 shows list of genes searched with term ‘NPHP’, ‘SLSN’, ‘JBTS’, ‘MKS’ and ‘Bardet-Biedl syndrome (BBS, syndrome of NPHP, RP, obesity, polydactyly, cognitive impairment, and male infertility)’, representatives of NPHP-RC.

NPHP-RC Genes
Among the known genes causing NPHP, a large deletion of NPHP1 is the most common, accounting for approximately one fifth of NPHP cases [16-20]. It is the first gene discovered as cause of NPHP, and it has homologous segments nearby, rendering this gene prone to large deletion (Fig. 4). Those with NPHP1 total deletion mostly have juvenile NPHP, whose onset of CRF is later than 5 years of age, with of ESRD about 13 yrs. Diagnosis of NPHP1 total deletion is rather straightforward, because multiplex PCR of NPHP1 exons does not produce band in NPHP1 total deletion. Recently, Korean patients with total NPHP1 deletion were reported to have unexpected findings of retinopathy with large or small flecks, compatible with Stargardt disease or albipunctatus retinopathy [21] (Fig. 5, 6); the authors suggested that children with impaired renal function of unknown cause should be screened for retinopathy, and retinopathy warrants screening for a homozygous deletion of NPHP1.

NPHP1 with nearby homologous segments, rendering this gene prone to large deletion. Courtesy from Dr. Hae Il Cheong.
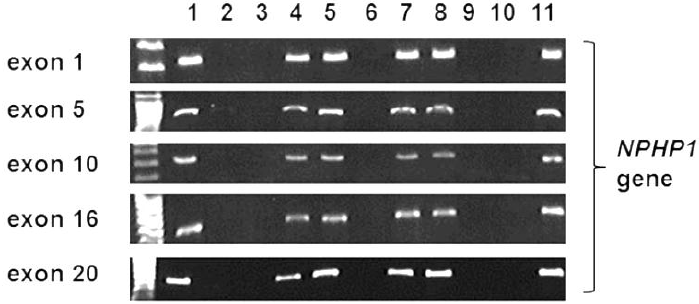
The results of amplification of the exons of NPHP1 by polymerase chain reaction, which revealed a failure of amplification (homozygous deletion) of all exons of NPHP1 in patients. (Lanes 1 and 11, control subjects; lanes 2, 3, 6, 9, and 10, patients with total deletion of NPHP1; lanes 4, 5, 7, and 8, family members of patients). Reprint with permission [21].
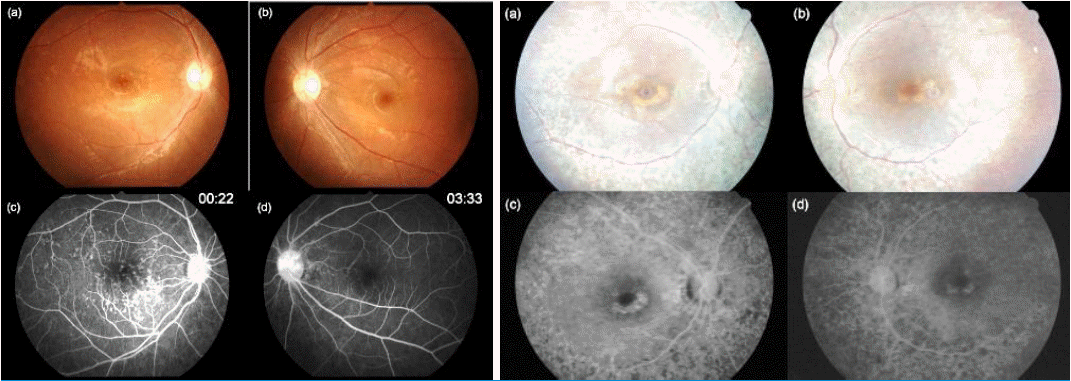
A fundus photograph and fundus fluorescein angiograms of the eyes of two Korean patients with total deletion of NPHP1. Left) (a) The fundus photograph shows a normal disc and yellow flecks on the posterior pole in the right and (b) left eyes. (c) A fluorescein angiogram 22s after dye injection shows multiple hyperfluorescent lesions corresponding to the flecks on the fundus photograph of the right eye. (d) A fluorescein angiogram 3.5min after dye injection shows no definite hyperfluorescence and choroidal silence in the left eye.
Right) (a) The fundus photograph shows a normal disc, a yellow fleck at the parafovea, and retinal pigment epithelium (RPE) degeneration along the major arcades without involvement of the far peripheral retinal area in the right (b) and left eyes. (c) A fluorescein angiogram 2min after dye injection shows a bull's eye appearance in the right eye. Maculopathy is shown with a foveal decrease in fluorescence surrounded by a continuous ring of increased fluorescence. Discrete areas of RPE atrophy (transmission window defect) surrounding the fovea (d). Reprint with permission [21].
Other genes contribute less than 2-3% [9]. Some of them are strongly associated to certain phenotype, NPHP5 or NPHP6 mutation lead to RP, WDR19 (NPHP13) is associated with Caroli disease [22] (Fig. 7). On the other hand many of them share phenotypes, demonstrated by the number of genes associated with each syndrome (Table 1). Due to the large number of genes to test, recently. next-generation sequencing (NGS) is often applied, and about 1/3 patients obtain genetic diagnosis, implying that there are yet many genes to discover [23].
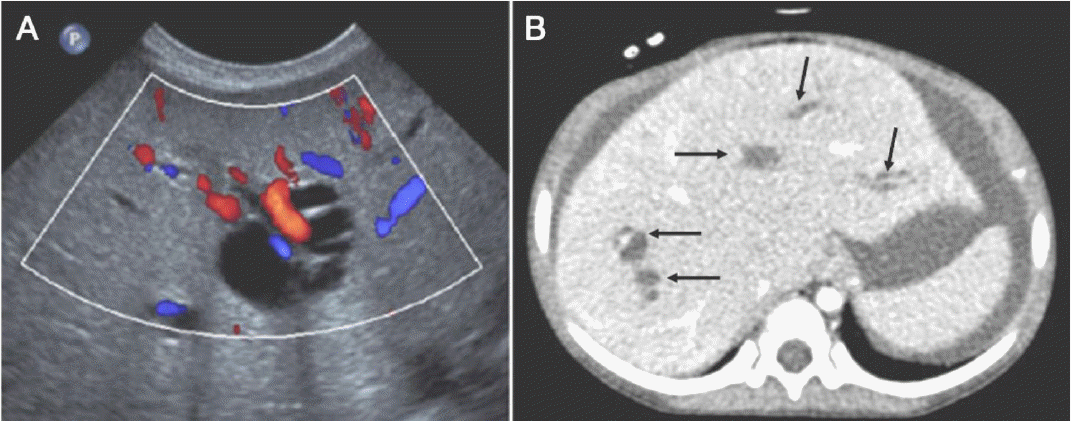
Liver images of patients with WDR19 mutations. (A) Abdominal Doppler ultrasonography reveals variable dilatation of the intrahepatic bile ducts in patient III-1. Red or blue tubular structures indicate hepatic vessels. (B) Axial computed tomography image of patient IV-1 shows globular enlargement of the liver and dilatation of the intrahepatic bile ducts (arrows), which are consistent with Caroli syndrome. Reprint with permission [22].
Pathogenesis
Primary cilia (Fig. 8) is present in almost all mammalian cells, functioning as sensory organelles, responding to flow, optic, osmotic, chemo or olfactory stimuli [24, 25], linking to various cellular function such as polarity, cell-cycle control [10]. Therefore defect of primary cilia of renal tubular cells results in cystic disease. Extra-renal symptoms are explained by presence of primary cilia in respective cells; Primary cilia at retina are photoreceptors, whose defect can cause retinopathy, oculomotor apraxia, nystagmus, and coloboma [10, 15, 26, 27]. Primary cilia in choangiocytes explain hepatic fibrosis in NPHP-RC [28]; Primary cilia in chondrocytes explains skeletal abnormalities such as short ribs, coneshaped epiphysis, and postaxial polydactyly in Jeune syndomre and JBTS or BBS [29, 30], especially with defect in genes of intra-flagella transport (IFT) [29].
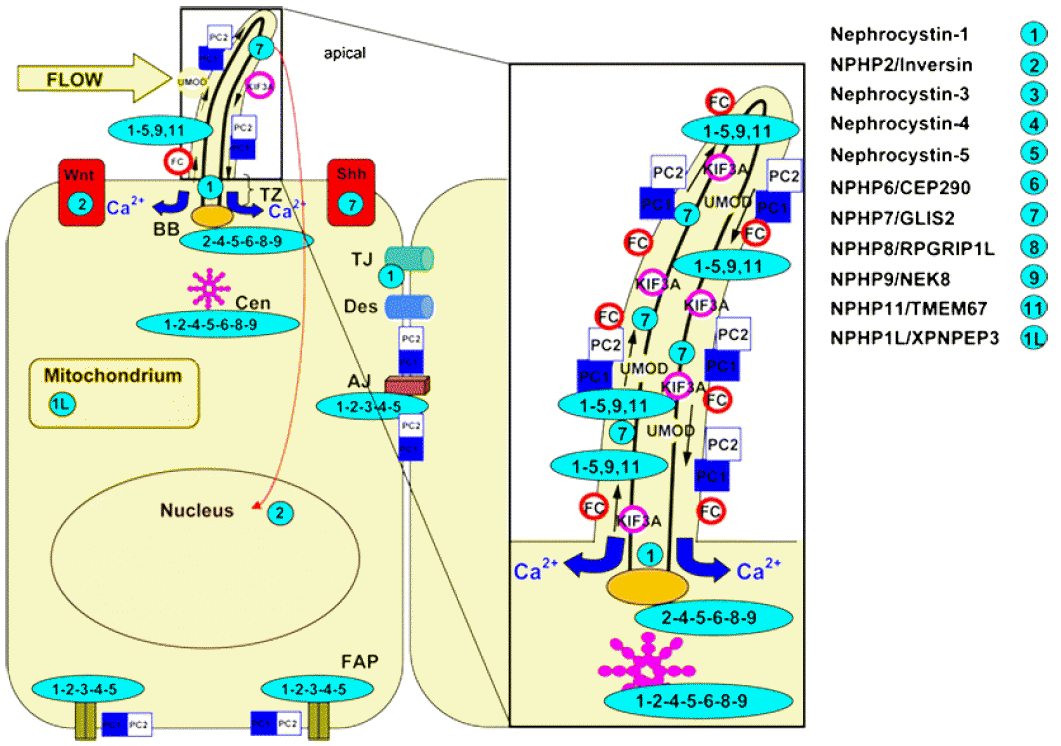
Subcellular localization of the NPHP molecules nephrocystins. Nephrocystins are detected in the primary cilia, basal bodies, the mitotic spindle, focal adhesions, and adherens junctions. Most nephrocystins are expressed in the primary cilium (PC, enlarged box), the basal body (BB), and centrosomes (Cen) in a cell cycle-dependent manner. NPHP1 is expressed in the transition zone (TZ), focal adhesion plaques (FAP), adherens junctions (AJ), and tight junctions (TJ). Arrows in the cilium show the directions of the anterograde and retrograde transport along the microtubule transport. The intraflagellar transport is mediated by kinesin 2, a heterotrimeric protein that is composed of two motor units (Kif3a and Kif3b) and one nonmotor unit (KAP3). Sensory cilia transfer external stimuli. Wnt and hedgehog (Shh) signaling interfere with planar cell polarity by affecting the orientation of the centrosomes and mitotic spindles. Reprint with permission [34].
Location, interacting molecules, and involved signaling pathway of respective causative genes are linked to the phenotype of various NPHP-RC. In addition, effect of additional mutations in another NPHP-RC genes or genetic modifiers has been suggested [9, 24, 31-33].
Treatment
There is no curative treatment for NPHP. Conservative management of CKD is necessary. There is no risk of recurrence after kidney transplantation; extra-renal symptoms progress irrespective of kidney transplantation.
Summary
NPHP is the most common monogenic cause of CKD in children or adolescents. Extra-renal symptoms often accompany, therefore examination of retina, hearing, and skeleton is necessary in patients with CKD with insidious onset. Genes involved in NPHP-RC are mostly related in primary cilia. While genetic diagnosis is necessary for definitive diagnosis, there is no curative treatment.
Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number : HI1 2C0014).”