Introduction
Fabry disease is a very rare X-linked genetic disorder arising from a deficiency in lysosomal ╬▒-galactosidase A, which catalyzes the breakdown of glycosphingolipid, especially globotriaosylceramide, which is present in the plasma membrane of most cells [1]. Insufficient enzyme activity results in the accumulation of globotriaosylceramide within the lysosomes of many cell types. Male patients are typically affected by corneal dystrophy, cutaneous involvement, neurologic abnormalities, and cardiovascular and renal diseases [2]. Atypical Fabry disease presents with oligosymptomatic manifestations, which are restricted to the heart and/or kidneys [3,4]. This X-linked disease is completely expressed in hemizygous males as a loss of ╬▒-galactosidase A, which is a definitive criterion for diagnosis. However, the clinical presentation is quite variable. Most female heterozygotes are considerably affected. However, unlike the typical descriptions, they show variable levels of enzyme activity, with a broader range of clinical symptoms [5,6]. Renal biopsy may be helpful in the diagnosis of Fabry disease, as intracellular vacuolization caused by intracellular lipid inclusions is noted in many cells, especially podocytes. Although unique intracellular osmiophilic structures, such as myeloid figures or zebra bodies, are highly characteristic of this disease, similar structures are detected in silicon nephropathy and iatrogenic phospholipidosis induced by various drugs, including amiodarone, chloroquine, and hydroxychloroquine [2,7-9]. Here, we report the case of a 10-year-old boy with hematuria who underwent a renal biopsy that showed the characteristic morphology of Fabry disease, even though the clinical findings were not consistent with this diagnosis.
Case report
A10.8-year-old boy presented with microscopic hematuria in a pediatric nephrology department. The microscopic hematuria was detected 3 years prior to this visit during a school urinary screening; however, the boy did not show any symptoms or signs. On admission, he was 150.1 cm tall (90-95th percentile) and weighed 42.0 kg (75-90th percentile). His blood pressure was 128/80 mmHg, and no specific findings were observed during systemic review and physical examination. He had neither a familial history of renal disease nor a history of medication use. Urine analysis yielded the following results: RBC, 10-30/HPF; protein -; occult blood, ++250; protein-to-creatinine ratio, 0.07; and calcium-to-creatinine ratio, 0.10. Renal function test results were normal (BUN/Cr, 13/0.86 mg/dL and eGFR, 155 mL/min).CBC, serum electrolytes, and blood biochemistry showed no abnormal findings. He was followed-up regularly for 3 years and was not prescribed any medication.
Renal biopsy was performed 3 years after the first visit. He still had microscopic hematuria without any other symptoms or signs. Based on the renal histologic findings, which suggested Fabry disease, ╬▒-galactosidase enzyme activity was measured. Enzymatic activity was in the normal range (normal range: 40-85 nmol/h/mg) for both the patient (41 nmol/h/mg) and his mother (45 nmol/h/mg). No mutation in the GLAgene was found. Criteria indicative of Fabry disease, such as acroparesthesia, angiokeratoma, sweating abnormalities, eye problems, stroke, and left ventricular hypertrophy, were not detected at follow-up.
A second renal biopsy was performed when he was 16 years old. He still had microscopic hematuria. No other abnormal symptoms were observed, and he showed normal ╬▒-galactosidase enzyme activity (50.8 nmol/h/mg).
Renal biopsy
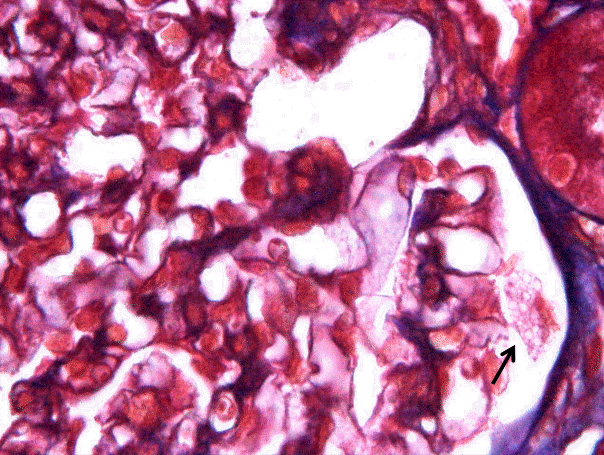
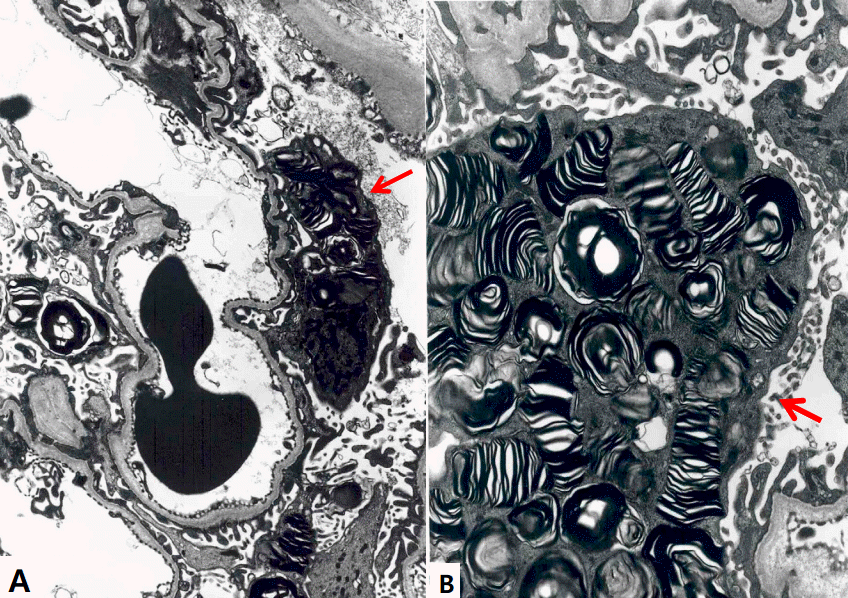
Two cores of renal cortex and medulla were obtained in the first renal biopsy, and 10 glomeruli were observed under light microscopy in paraffin-embedded sections. The glomeruli were normocellular and normal in size. The mesangium was not expanded. The visceral epithelial cells in one glomerulus had plump, vacuolated cytoplasm in 3 out of 10 glomeruli and one or two podocytes were showing this zebra body in one glomerulus (Fig. 1); however, the other glomeruli were unremarkable. The glomerular basement membrane was not thickened. The tubules and interstitium were well preserved, and none of the cells had vacuolar cytoplasm. The blood vessels showed no evidence of sclerosis or vasculitis. Immunofluorescence staining for immunoglobulin (Ig)G, IgA, IgM, complement (C)3, C4,and fibrinogen were negative.
In the ultrastructural examination, we observed that the podocytes contained prominent membrane-bound, electron-dense bodies showing a multilamellar appearance, characteristic of myelin figures or zebra bodies (Fig. 2A and B). The curvilinear bodies were not detected. The glomerular basement membrane was of normal thickness, and the foot processes showed focal effacement. No immune-type electron-dense deposits were noted.
In the second renal biopsy, one core of the renal cortex, which contained 13 glomeruli, was obtained. No diagnostic abnormalities were observed under light microscopy and by immunofluorescence staining. On ultrastructural examination, membrane-bound, electron-dense bodies were not detected in the glomeruli, tubules, or vessels. The thickness of the glomerular basement membrane was normal (mean, 336.58 nm).
Discussion
The renal histologic findings of Fabry disease were first described about 50 years ago [10,11]. In Fabry disease, lysosomal ╬▒-galactosidase A deficiency leads to enlargement of glomerular podocytes with clear cytoplasmic vacuoles observable under light microscopy, caused by the accumulation of globotriaosylceramide in the lysosome. Ultrastructural examination of the renal tissue in Fabry disease shows typical inclusion bodies that look like ŌĆ£onion skinŌĆØ with a ŌĆ£zebraŌĆØ appearance owing to the concentric lamellation of transparent and dark layers in the cytoplasm of all renal cell types. These characteristic structures are found in tubular epithelial cells; all glomerular cells, especially podocytes; and the endothelial cells of renal vessels. In our case, immunofluorescence staining for immunoglobulins and complement components was negative. Electron microscopic examination revealed abnormal findings in the glomerular basement membrane, such as duplication, although these findings are rare [12]. These are prominent characteristics of Fabry disease; however, they are not diagnostic. Therefore, differential diagnosis to rule out other diseases showing lipid accumulation in lysosomes is important. In this case, there were neither clinical symptoms nor signs of Fabry disease. Alpha-galactosidase A levels in the leukocytes were normal, and no mutation in the GLA gene was detected;however, zebra bodies were observed in the first renal biopsy by electron microscopy. There was no familial history of hematuria or Fabry disease. The most useful finding during differential diagnosis is the location and shape of the multilamellated myelin figures [9]. Diseases showing lipid accumulation in glomerular podocytes, similar to Fabry disease, include infantile nephrosialidosis, GM1 gangliosidosis, I-cell disease (mucolipidosis, type 2), Hurler syndrome, and Niemann-Pick disease. However, among these diseases, only Niemann-Pick disease shows distinct myelin figures [8]. Iatrogenic phospholipidosis shows histologic features that are most similar to those of Fabry disease [9], and it is caused by several drugs, including chloroquine, amiodarone, and aminoglycosides [8,9,13,14]. Chloroquine was developed as a therapeutic agent for malaria and amebiasis, and it is also used in the treatment of autoimmune diseases such as rheumatoid arthritis and lupus erythematosus. Chloroquine has serious adverse effects, including renal toxicity, ophthalmic keratopathy, dermatopathy, and gastrointestinal and neurologic symptoms. One study showed that renal function of patients with Sj├ČgrenŌĆÖs syndrome was worse in those who consumed chloroquine, and they had progressive renal failure, although it was dose independent. After cessation of drug therapy, renal function recovered, and the myeloid bodies seen in the podocytes disappeared [2]. In iatrogenic phospholipidosis caused by chloroquine and hydroxychloroquine, curvilinear bodies (CLB) were also detected in the podocytes and vascular smooth muscle cells. This is a useful finding for the differential diagnosis of Fabry disease. The pathogenesis of chloroquine-induced iatrogenic phospholipidosis is not clear; however, chloroquine is an amphophilic, lysosomotropic substance that is similar to many other pharmacologically unrelated drugs (e.g., amitriptyline, chlorpromazine, and gentamycin). Therefore, lysosomal damage is likely to be an important step in chloroquine toxicity [15]. Chloroquine is a weak base that crosses the lysosomal membrane and becomes concentrated therein by protonation. The resulting increase in lysosomal pH is an important mechanism in the inactivation of lysosomal enzymes and impairment of degradative capacity [16,17].
The patient in our case had no history of medication use prior to renal biopsy. We could not find any other similar case in the literature. This is a very rare case that resulted from the temporary accumulation of normal or abnormal substances in the lysosome due to an unknown cause. Moreover, lysosomal inclusion bodies (zebra bodies or myelin figures) are not specific to the diagnosis of lysosomal storage diseases such as Fabry disease. Therefore, in addition to a renal biopsy and clinical history evaluation, a leukocyte ╬▒-galactosidase A enzyme assay and genetic testing for a GLA mutation are essential to confirm the diagnosis of Fabry disease because lysosomal inclusion bodies, similar to those observed in this patient, can be caused by numerous and unknown conditions.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print