Introduction
C3 glomerulopathy (C3G) is a recently defined pathological entity characterized by C3 accumulation with absent or scant immunoglobulin deposition leading to variable glomerular inflammations [1,2]. C3G was often diagnosed previously as membranoproliferative glomerulonephritis (MPGN) because of similarities in the light microscopy (LM) and electron microscopy (EM) findings. MPGN can be classified into either immunoglobulin (IgG)-associated MPGN (caused by classical complement pathway activation); MPGN with dominant C3 (C3G and C3GN); or idiopathic MPGN. Both C3GN and idiopathic MPGN are caused by alternative complement pathway activations [3].
C3G includes C3 glomerulonephritis (C3GN) and dense deposit disease (DDD) [4,5]. The difference between C3GN and DDD is that DDD is characterized by the presence of extremely electron-dense deposits in the basement membrane, while the changes observed in patients with C3GN are more heterogenous. C3GN also includes disease entities associated with complement mutation, which is causally associated with the underlying renal pathology, such as familial DDD with C3 mutation and familial C3GN with mutations in the CFHR genes. The incidence of C3GN is reported to be one to two per 1,000,000 children, with its onset occurring between 5 and 15 years of age. The clinical manifestations of C3GN comprise a wide spectrum, including mild urinary abnormalities, nephrotic syndrome, and nephritic syndrome with or without renal impairment [1,3,6,7]. The condition often progresses to chronic kidney disease or end-stage kidney disease and can recur after renal transplantation.
In affected patients, the alternative pathway produces C3 convertase, which amplifies C3 activation, resulting in the creation of C3b particles, and finally, the formation of C5 convertase to assemble MAC C5b-9. This damages the cells via the development of a lytic pore and leads to cellular death via apoptosis or necrosis. Factor H and other complement proteins regulate the amplification of C3 activation [8].
The current treatment includes supportive measures and use of immunosuppressants, including prednisone, mycophenolate mofetil (MMF), and calcineurin inhibitors. Recently, plasma exchange/plasma infusion and provision of eculizumab, a monoclonal antibody against C5, can be used in cases of nephritic syndrome and/or decreased renal function in patients with C3GN.
Case report
Proteinuria and microscopic hematuria were first detected via a school urinary screening in a 10-year-old girl. The patient was referred to our hospital for evaluation by a local hospital. Her physical examination findings were normal, there was also no edema. The patient had a normal blood pressure of 100/60 mmHg. The laboratory investigation results upon admission were as follows: leukocyte count, 9,170/╬╝L; platelet count, 460 K/╬╝L; hemoglobin level, 12.8 g/dL; BUN level, 11.9 mg/dL; and serum creatinine level, 0.7 mg/dL (eGFR, 94.7 mL/min/1.73 m2, Schwarz formula). Urinalysis showed only microscopic hematuria (RBC count, 20-30/HPF) by dipstick, and the level of protein identified in the 24-hour urine study was 47 mg/m2/day.
A markedly decreased C3 level of 15 mg/dL (reference range: 77-195 mg/dL) and normal C4 level of 15.9 mg/dL (reference range: 7-40 mg/dL) were noted on immunological evaluation. The infectious serology tests conducted for hepatitis B and hepatitis C revealed negative findings. Renal imaging showed that the patient had normal-sized kidneys, with ureteral duplication. Renal biopsy was performed owing to persistently decreased C3 level. LM showed diffuse exudative proliferative glomerulonephritis. On immunofluorescence staining, a markedly increased granular deposit of C3 in the mesangium was noted without any other immunoglobulin deposition. EM revealed an electron- dense deposit along the glomerular basement membrane. We initiated the administration of angiotensinconverting enzyme (ACE) inhibitors for supportive care. The patient showed only microscopic hematuria and decreased serum C3 level without proteinuria for -3 years. However, we subsequently failed to follow up with the patient, and after -2 years, the patient revisited our hospital with nephrotic-range proteinuria (53.37 mg/m2/h) and microscopic hematuria with normal renal function at the age of 15 years. She reported no symptoms during the entire period. A second biopsy was performed, which showed an MPGN pattern with predominant C3 deposits on immunofluorescence staining, similar to those observed on the first biopsy (Fig. 1).
At this time, the patientŌĆÖs levels of complement factor H, factor B, and anti-CFH autoantibody were within normal ranges. However, the level of complement factor I (CFI) decreased to 0.47 mg/dL, as compared with the reference value (3.8-5.8 mg/dL). There was no mutation of CFI by Sanger sequencing noted. During the genetic study for the alternative pathway of the complement system, a heterozygous mutation was detected in a C8A gene mutation known to be associated with atypical hemolytic uremic syndrome (aHUS) or DDD. However, the C8╬▒ plasma level and hemolytic activity were needed to confirm the presence of a meaningful mutation of the C8A gene.
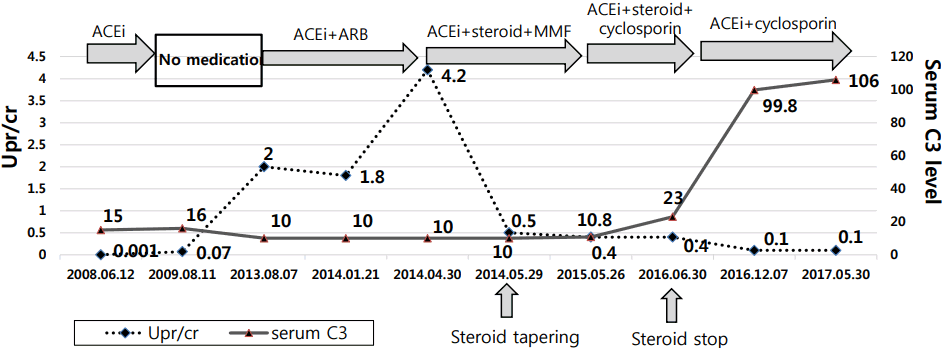
We started to administer ACE inhibitors and angiotensin receptor blockers (ARBs) to reduce the proteinuria. Thereafter, the amount of proteinuria was decreased with these treatments; however, after -8 months, proteinuria was increased to 95 mg/m2/h. At that time, it was decided that we could not use eculizumab because it was expensive; thus, we added oral prednisolone (40 mg/day) and MMF (1 g/day). Following prescription, proteinuria was decreased to UPr/ cr of 0.4; however, proteinuria was sustained with a consistently low serum C3 level (10.8 mg/dL, reference range: 77-195 mg/dL). MMF was replaced with cyclosporine. It was also determined that we could not use prednisolone for long-term maintenance because the patient demonstrated a cushingoid appearance and amenorrhea. Thus, we tapered off the dosage of prednisolone and decided only to use cyclosporine and an ACE inhibitor for maintenance. After 6 months, the patientŌĆÖs proteinuria had improved, and her serum C3 level recovered back to normal (106 mg/dL, reference range: 77-195 mg/dL). We continued to administer cyclosporine and ACE inhibitor; thereafter, we attempted to taper off the dosage of cyclosporine. However, when we reduced the dosage of cyclosporine, her proteinuria worsened; hence, we continued to administer cyclosporine (150 mg/day) and ACE inhibitor (Fig. 2).
Discussion
We reported a case of C3GN detected via school urine examination in a 10-year-old girl, which was confirmed via renal biopsy and showed a membranoproliferative pattern with electron-dense deposits on the subendothelium and interstitial fibrosis. A low serum C3 level is commonly observed in patients with this disease, but does not correlate with the severity of the condition [9]. The prognosis of these patients may be determined by a decrease in the eGFR and the severity of proteinuria at the time of diagnosis and during follow-up as in most renal diseases [10]. The degree of tubulointerstitial fibrosis is another poor prognostic factor, more so than the severity of glomerular changes [11]. There is no consensus regarding recommendation of a specific treatment in children with C3GN. There is also no evidence for the treatment with ACE inhibitors or ARBs. However, these medications should only be considered for use because of the existing ability to extrapolate information from other proteinuric renal diseases. The limited data on the use of ACE inhibitors or ARBs in patients with C3GN had been reported in a French C3G cohort, where it was stated that treatment with ACE inhibitors or ARBs might improve renal survival (P<0.0001) [12].
Some studies have reported that anticellular immune suppression is considered less effective in children with C3GN. Servais et al. reported that immunosuppressants were not associated with renal survival [12]. Another previous study reported the failure of treatment with glucocorticoids in helping patients achieve remission after 5 years [13].
However, recent case series suggested that the administration of MMF is effective [14,15]. Furthermore, a recent KDIGO (Kidney Disease: Improving Global Outcomes) controversies conference recommended that C3G patients with moderate disease (defined as urine protein of more than 500 mg/24 hours or recent rise in creatinine or moderate inflammation on renal biopsy) despite supportive therapy receive prednisone or MMF [16]. There are also reports for the effectiveness of cyclosporin in patients with C3G. Bagheri et al. noted that 18 patients with MPGN who were treated with cyclosporine showed an excellent response to the medication [17].
Recently, new therapies have been suggested to target complement pathways owing to an improvement in the understanding of the pathogenesis of C3GN. An anti-complement effect for C3G has been predicted via the use of an animal model [18]. C3G complement blockade eculizumab is a monoclonal antibody targeted against complement C5 that inhibits the activation of the alternative complement pathway. Although reported results to date are conflicting, many studies have reported an efficacy of eculizumab in patients with C3G, even in children [18,19]. Eculizumab is approved by the United States Food and Drug Administration for the treatment of aHUS, thrombotic microangiopathy, and paroxysmal nocturnal hemoglobinuria. Therefore, it is used as an off-label medication for C3G.
As mentioned above, recently conducted studies explain the effectiveness of eculizumab because of the understanding of the pathogenesis of C3G. However, eculizumab cannot be used in some instances, and in these instances, it has been reported that cyclosporine is effective for managing steroid-resistant nephrotic syndrome caused by MPGN II [20-22]. Our patient was partially responsive to MMF treatment but had persistent proteinuria and a low serum C3 level. When we switched MMF to cyclosporine, her proteinuria resolved, and the serum C3 level also normalized. Therefore, we reported a case of C3GN successfully treated with cyclosporine. We suggest that cyclosporine be considered as a therapeutic agent for C3GN in cases in which eculizumab cannot be used and/or if the patient does not respond to other immunosuppressants, such as steroids and MMF.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print