Introduction
Amyloidosis is a clinical disorder caused by the deposition of an extracellular insoluble protein in various body tissues, which causes progressive organ dysfunction. Amyloidosis can be classified into primary or secondary types, and the deposition of amyloid fibrils can be localized or systemic [1]. Clinical manifestations of amyloidosis depend upon the involved organs and the amount of amyloid fibril deposition [2].
Secondary amyloidosis predominantly affects the kidneys, and renal involvement is the most important predictor of survival in patients. Clinical manifestations of renal amyloidosis include proteinuria, nephrotic syndrome, renal insufficiency and progression to end-stage renal failure [1,2]. Inflammatory bowel disease (IBD) is a chronic inflammatory condition of the gastrointestinal tract that includes a diverse group of complex and multifactorial disorders [3]. Secondary renal amyloidosis is an uncommon but serious complication of IBD and has primarily been reported in adults with CrohnŌĆÖs disease. Amyloidosis develops in approximately 1% of patients with CrohnŌĆÖs disease during the course of the disease and is a leading cause of death with a longterm mortality rate of 40ŌĆō60% in this patient population [4,5]. A few case reports have described amyloidosis in pediatric patients with IBD.
We report a pediatric case of IBD complicated with renal amyloidosis. This study was approved by the Seoul National University Hospital's Institutional Review Board (IRB No. 1808-130-967).
Case report
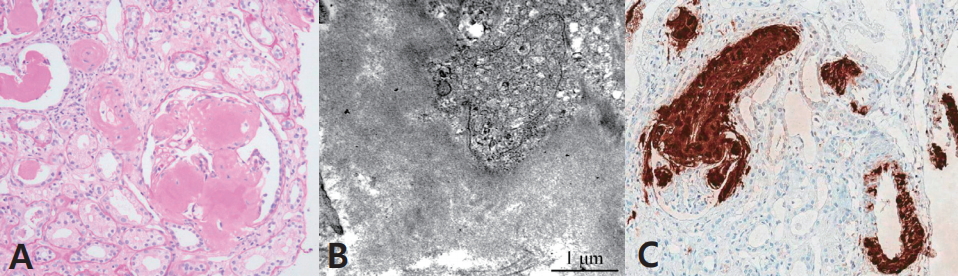
A 19-year-old young man with a long-term history of IBD was referred to the Division of Nephrology, Department of Pediatrics, Seoul National University ChildrenŌĆÖs Hospital, Seoul, Korea with a high serum creatinine level (1.55 mg/dL). The patient reported history of rectal bleeding at 3 years old age, and he was tentatively diagnosed with an anal fissure and constipation. A year later, he developed bloodtinged stool and abdominal pain. Clinical, laboratory, and colonoscopic examinations at our hospital led to a diagnosis of colonic angiodysplasia and iron deficiency anemia. He was managed with blood transfusion and iron supplementation. A year thereafter, he developed bilateral knee joint pain with swelling and tested positive for the perinuclear anti-neutrophil cytoplasmic antibody. Colonoscopic examination showed severe colitis throughout the colon, and a colonic biopsy revealed chronic active colitis without granuloma formation. He was diagnosed with ulcerative colitis and treated with oral azathioprine, mesalazine, and prednisolone for 5 years. At 10 years of age, his family moved to their home-town, and he was followed-up at a local hospital and received only mesalazine. At 15 years of age, he was admitted to that hospital with new-onset abdominal pain and vomiting. Abdominal computed tomography revealed a segmental polypoid mass at the sigmoid colon, and he was referred to our hospital for further evaluation of the mass. At our hospital, he underwent total proctocolectomy with ileal pouch-anal anastomosis. Histopathological examination of the mass revealed stage II moderately differentiated sigmoid colon cancer. Postoperatively, he experienced persistent abdominal pain with diarrhea and weight loss over the subsequent 6 months. Follow-up gastrointestinal endoscopy revealed features of CrohnŌĆÖs disease involving the upper gastrointestinal tract, as well as the small bowel and the colon. Granulomas were detected in all 7 biopsy specimens examined. He was treated with intravenous methylprednisolone and oral azathioprine followed by 18 infusions of infliximab (200 mg/dose) administered every 6 weeks, which led to improvement in his gastrointestinal symptoms and inflammatory parameters. Thereafter, he was lost to follow-up for 2 years. At 18 years of age, he was admitted to another hospital with acute abdominal pain. He was treated with 3 infusions of infliximab followed by 12 infusions of adalimumab over 9 months followed by the administration of oral corticosteroid and azathioprine. However, he was re-referred to our hospital owing to persistent diarrhea with severe abdominal pain. Infliximab infusion was re-initiated concomitant with oral azathioprine and rifaximin. A month later, he was referred to the Division of Nephrology owing to new-onset azotemia. He was 153 cm tall (<3rd percentile) and weighed 37.4 kg (<3rd percentile). Physical examination was unremarkable except for his poor general condition. Laboratory tests revealed his hemoglobin was 9.1 g/dL, serum creatinine level was 1.55 mg/dL, estimated glomerular filtration rate (eGFR) calculated using the Schwartz formula was 40.6 mL/min/1.73m2, serum albumin level was 2.7 g/dL, and C-reactive protein was 9.9 mg/dL. Serological tests including serum complement C3 and C4, anti-nuclear antibody, antineutrophil cytoplasmic antibody, and anti-double stranded DNA were all negative. Urinalysis showed 3+ proteinuria and microscopic hematuria, and 24-hour urinary protein excretion was 5.9 g/day. Renal ultrasonography showed bilateral globular kidneys with increased renal parenchymal echogenicity and splenomegaly. Renal biopsy revealed deposits of amorphous pinkish material in the mesangial matrix and vascular walls (Fig. 1A). Electron microscopic examination showed glomerular and perivascular deposition of non-branching amyloid fibrils (Fig. 1B). Immunofluorescence microscopy was strongly positive for amyloid (Fig. 1C). The serum amyloid A (SAA) protein level was 13.4 ╬╝g/dL (reference range <8 ╬╝g/dL). These findings were consistent with secondary renal amyloidosis. He was treated with oral prednisolone, mesalazine, methotrexate, and repeated infliximab infusions. After the 6th infliximab infusion, clinical parameters of IBD showed significant improvement, although renal dysfunction continued to progress (serum creatinine 2.46 mg/dL and eGFR 31.1 mL/min/1.73m2).
Discussion
Amyloidosis is a single- or multiorgan disease caused by the extracellular deposition of low-molecular weight, insoluble, rigid, and amorphous proteinaceous material. To date, 25 different proteins causing amyloidosis have been identified. The formation of amyloid commences with the misfolding of amyloidogenic precursor proteins followed by self-aggregation and the formation of protofilaments from the misfolded proteins. Amyloid fibrils comprise 4ŌĆō6 protofilaments showing a ╬▓-pleated sheet configuration and produce birefringence under polarized light when stained using the Congo red dye. Amyloid deposition causes structural and functional impairment of the affected tissues and organs [5,6].
Amyloidosis can be classified on the basis of: 1) the nature of the precursor proteins or, 2) the distribution of amyloid deposits. Localized amyloidosis involves the local production and deposition of an amyloidogenic protein (commonly immunoglobulin light chains) within the affected organ. Systemic amyloidosis involves the deposition of circulating amyloidogenic proteins in various organs. Primary amyloidosis (amyloid light-chain [AL] amyloidosis), the most common type, is characterized by the deposition of excess immunoglobulin light chains in patients with multiple myeloma, lymphoma, or other lymphoproliferative diseases. Secondary amyloidosis (amyloid A [AA] amyloidosis) is caused by the deposition of N-terminal fragments of SAA proteins in patients with chronic inflammatory, infectious, or neoplastic diseases. SAA is an acute phase reactant protein, which is produced during sustained inflammatory conditions [7].
Secondary amyloidosis is a rare but serious complication of cancers, chronic infections, or chronic inflammatory disorders including IBD, particularly CrohnŌĆÖs disease [7]. The underlying inflammatory activity is an important risk factor for the development and progression of secondary amyloidosis. Amyloidosis develops in patients with IBD showing uncontrolled prolonged inflammatory activity. In our patient, amyloidosis developed 14 years after the onset of poorly controlled IBD.
Diagnosis of renal amyloidosis is confirmed by histopathological examination showing amyloid deposition within tissues. Light microscopic examination using hematoxylin and eosin stains shows amorphous material within the mesangium and the glomerular capillary loops. The gold standard of diagnosis is Congo red staining of amyloid, showing apple-green birefringence under polarized light. Electron microscopic examination reveals non-branching amyloid fibrils [5,9].
Our patient reported a long-term history of IBD with a complicated disease course. The earlier part of the case with ulcerative colitis and sigmoid colon cancer was reported previously [10]. The patient was diagnosed with ulcerative colitis at 5 years of age and CrohnŌĆÖs disease at 15 years of age. Ulcerative colitis and CrohnŌĆÖs disease are the two representative forms of IBD showing multifactorial pathogenesis and are characterized by chronic, remittent gastrointestinal involvement. The two conditions are often indistinguishable owing to overlapping initial histopathological features. In our patient, the presence of granulomata and small bowel involvement manifested later in the course of the disease resulted in a change of diagnosis to CrohnŌĆÖs disease.
The Pediatric Modification of the Montreal Classification for IBD: The Paris Classification was proposed in 2011 [11]. This classification defines IBD occurring in children aged <6 years as ŌĆ£very early onset IBDŌĆØ, which differs from IBD diagnosed in older children [11,12]. This subtype of IBD accounts for approximately 15% of all childhood-onset IBD cases [3]. Our patient was classified as a case of very early onset IBD.
While IBD primarily affects the gastrointestinal tract, extraintestinal manifestations occur in approximately 25 % of pediatric patients [13], and the biliary tract, the joints, skin, and the eyes are commonly involved. Renal and urinary tract involvement is rare and includes kidney stones, enterovesical fistulas, ureteral obstruction, and renal amyloidosis [14]. Our patient showed no extraintestinal complications except renal amyloidosis.
Secondary amyloidosis primarily affects the kidneys, and common clinical manifestations are proteinuria, nephrotic syndrome, and renal insufficiency. Proteinuria is more common than renal insufficiency at the time of diagnosis [15]. A characteristic finding of renal amyloidosis is bilaterally increased renal mass secondary to amyloid deposition, as demonstrated by renal ultrasonography performed in our patient.
Early diagnosis of renal amyloidosis in patients with IBD may prevent further loss of organ function [14]. The goal of treatment is to strict control of the underlying inflammatory disease and therefore reduced production of SAA protein [9,14,16]. Monitoring serum levels of the SAA protein is vital to assess adequate suppression of underlying disease [5]. Several reports suggest that intensive therapy for IBD patients induced rapid decreased in proteinuria, but renal function was not completely recovery [14,17,18].
In conclusion, we report a case of secondary renal amyloidosis in a pediatric patient who reported a 16-year history of ŌĆ£very early onset inflammatory bowel diseaseŌĆØ. Intensive treatment including repeated infliximab infusions improved clinical parameters of inflammatory bowel disease, although renal dysfunction showed progression. Amyloidosis should be considered in patients with IBD, particularly if they suffered disease progression.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print