Introduction
Primary hyperoxaluria (PH) is a rare autosomal recessive inborn error of glyoxylate metabolism that results in endogenous overproduction of oxalate [1]. PH is a genetically and clinically heterogeneous disease and is classified into three subgroups.
PH type 1 (PH1), the most frequent and severe subgroup accounting for the majority of cases, is caused by mutations in AGXT, which encodes the peroxisomal liver-specific enzyme alanine-glyoxylate aminotransferase (AGT) [2]. AGT catalyzes the conversion of glyoxylate to glycine. Defective function of AGT results in conversion of glyoxylate to oxalate, which accumulates in the kidneys and other organs by forming insoluble calcium oxalate salts. Progressive renal deposition of calcium oxalate results in urolithiasis and/or nephrocalcinosis, followed by renal failure. Excessive blood levels of oxalate may lead to systemic oxalosis with oxalate deposition predominantly in the bone, skin, retina, myocardium, vessel walls, and the central nervous system. PH type 2 (PH2) is caused by mutations in GRHPR , which encodes glyoxylate reductasehydroxypyruvate reductase (GRHPR) [3]. GRHPR catalyzes the reduction of glyoxylate to glycolate. Functional deficiency of GRHPR results in abnormally increased metabolism of glyoxylate to oxalate by lactate dehydrogenase. PH type 3 (PH3) is caused by mutations in HOGA1 , which encodes the liver-specific mitochondrial enzyme 4-hydroxy-2-oxoglutarate aldolase (HOGA) [4]. It is unclear why defective HOGA causes oxalate accumulation.
Combined liver and kidney transplantation is the treatment of choice in patients with PH1, whereas isolated kidney transplantation is recommended for patients with PH2. Neither kidney nor liver transplantation is required in patients with PH3, as none of the patients with PH3 have been reported to develop end-stage renal disease (ESRD) [5].
Although there have been several case reports of PH in Korea [6,7], systematic studies have not been reported yet. Herein we report 11 cases of Korean patients with PH, with retrospective analyses of genotype-phenotype correlations and the outcome of combined liver and kidney transplantation in five patients.
Materials and methods
In total, 11 pediatric patients who were diagnosed with PH during the period from December 2008 to July 2018 at three tertiary care centers (Seoul National University ChildrenŌĆÖs Hospital Seoul, Korea, Asan Medical Center, Seoul, Korea, and Pusan University ChildrenŌĆÖs Hospital, Yangsan, Korea) were recruited. All patients were non-consanguineous Koreans, except for one patient (Patient 8) who had a Korean mother and Taiwanese father. Diagnosis of PH was based on increased urinary excretion levels of oxalate [5] and/or presence of genetic mutations in one of the three candidate genes, i.e., AGXT for PH1, GRHPR for PH2, and HOGA1 for PH3. Genomic DNAs of the patients and their available family members were isolated from their peripheral blood leukocytes. Entire coding regions of the three genes were amplified via polymerase chain reaction and sequenced directly. All the primers for polymerase chain reaction were designed to start from intronic sequences, and the primer sequences are available upon request.
1. Ethics statement
This study was approved by the Institutional Review Board of Seoul National University Hospital (IRB No. 0812-002-264). The patients and/or their parents provided informed consent to participate in this study.
Results
1. Genotypes
Mutational analyses revealed compound heterozygous or homozygous mutations in AGXT in nine patients. The remaining two patients carried a single heterozygous mutation in GRHPR and HOGA1 , respectively. Among 10 different AGXT mutations, 6 were truncating mutations. The c.33dupC mutation in AGXT was common with an allele frequency of 8/18 (44.4%) (Table 1).
2. Phenotypes
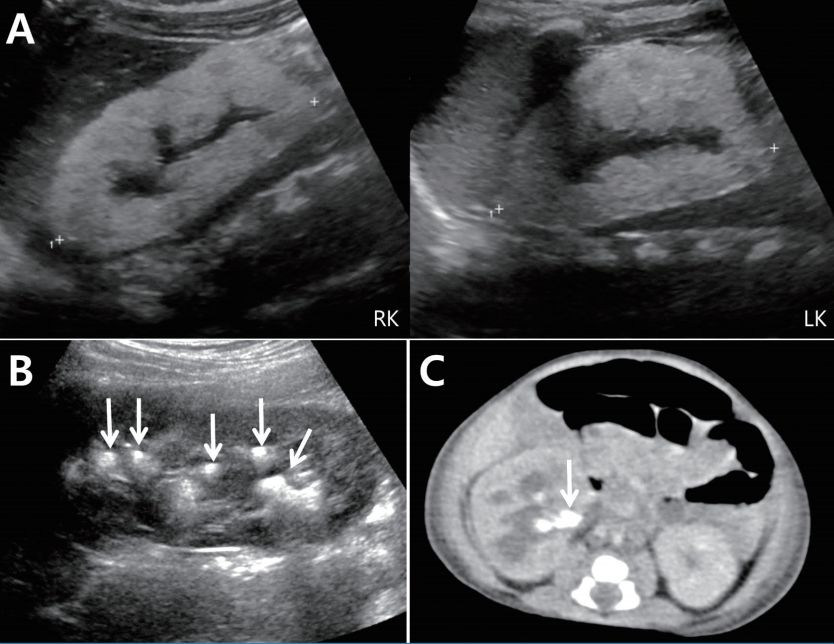
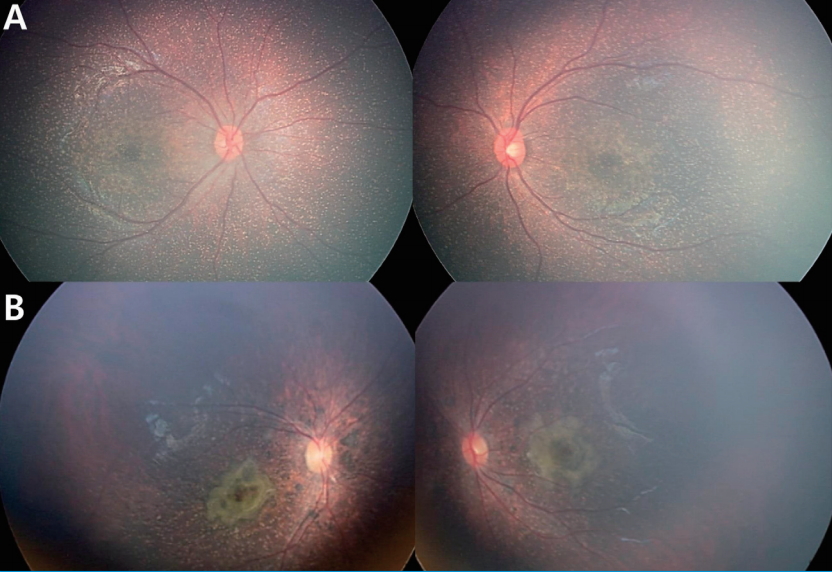
The clinical features and disease courses of the patients are summarized in Table 2. Among 11 patients in this study, 10 patients were boys and one patient with AGXT mutation was a girl. The median age of onset was 3 months (range, 2 monthsŌĆō3 years), and eight patients developed renal symptoms at the age of 5 months or younger. Eight patients with PH1 already had ESRD at initial presentation, and three patients presented with nephrolithiasis with/without gross hematuria. A family history of renal diseases was positive in two patients: Patient 7 had a grand-uncle who had ESRD of unknown causes, and Patient 9 had a grand-uncle who had an episode of spontaneous passage of a urinary stone. Urinary oxalate excretion levels were higher in two non-PH1 patients than in patients with PH1. This difference may be caused by different renal functions between both groups of patients. Nephrocalcinosis was detected in all patients on kidney ultrasonography and/or CT scan., except for Patient 9 who presented with nephrolithiasis, and nephrolithiasis was accompanied in five patients (Fig.1). Six patients with PH1 had retinal involvement (Fig.2), indicating systemic oxalosis.
3. Genotype-phenotype correlations
Among the nine patients with PH1, five patients (Patients 1, 3ŌĆō5, and 7) had truncating mutations in both alleles, and four patients (Patients 2, 6, 8, and 9) had one truncating mutation in one allele and one missense mutation in the other allele. All patients with two truncating mutations showed early onset ages (Ōēż5 months of age), whereas two of four patients with one truncating mutation showed older ages of onset at 2 years and 3 years, respectively. Retinal involvement indicating systemic oxalosis was detected in all patients with two truncating mutations, but in two of four patients with one truncating mutation. All patients with PH1 had ESRD at onset, except for Patient 9 who had one truncating mutation and presented with recurrent nephrolithiasis without renal functional impairment.
4. Disease courses
In four patients (Patients 1ŌĆō3, and 7), pyridoxine therapy was initially started with dialysis, but was discontinued after the genetic diagnosis, which showed that their genotypes were expected to indicate pyridoxine non-responsiveness.
Among the eight PH1 patients who presented with ESRD, five patients were initially treated with combined hemodialysis and nocturnal peritoneal dialysis to maximize oxalate removal, and three patients were treated with hemodialysis or peritoneal dialysis. Later, five patients (Patients 1ŌĆō5) underwent liver transplantation with/without subsequent kidney transplantation (n=3). Patient 1 underwent deceaseddonor liver transplantation at the age of 4 months. However, he developed fulminant sepsis due to surgical wound problems and died on the 51st postoperative day. Patient 2 underwent deceased-donor liver transplantation at the age of 6 years but developed primary graft non-function. He received a second liver transplantation from his mother soon thereafter. He is currently waiting for kidney transplantation while on hemodialysis. Patient 3 received deceased-donor liver transplantation at the age of 3 years followed by a living-donor kidney transplantation from his mother 3 months later. However, the graft kidney was lost due to primary graft non-function and again underwent a second kidney transplantation from a deceaseddonor. Currently, he has chronic kidney disease (CKD) stage 3 without dialysis. Patient 4 received deceased-donor liver transplantation at the age of 1 year followed by a deceased-donor kidney transplantation at the age of 5 years. Currently, his estimated glomerular filtration rate (eGFR calculated using the Schwartz formula) is 90.3 ml/min/1.73 m2. Patient 5 received deceased-donor liver transplantation followed by a deceased-donor kidney transplantation at the age of 6 months and 2 years, respectively. Currently, he has CKD stage 2. Among the remaining four patients with PH1, three patients (Patients 6ŌĆō8) are waiting for organ transplantation while on dialysis. Patient 9 maintains normal renal function although he had recurrent episodes of nephrolithiasis despite oral potassium citrate medication.
Patient 10 with non-PH1 presented with gross hematuria on the diaper at the age of 2 years. Kidney ultrasonography showed medullary nephrocalcinosis with multiple renal stones. He was prescribed high fluid intake and oral potassium citrate, and the latest kidney ultrasonography showed persistent medullary nephrocalcinosis without recurrence of nephrolithiasis. Patient 11 with non-PH1 presented with a staghorn stone in the right kidney with hydrouretronephrosis. He underwent percutaneous nephrostomy, and chemical analysis revealed that the stone was composed of 80% calcium oxalate, 10% carbonate apatite, and 10% struvite. Nephrolithiasis did not recur on oral potassium citrate medication and his renal function is maintained normally.
Discussion
This study showed a case series of 11 Korean pediatric cases, including nine cases with a genetic diagnosis of PH1. The estimated prevalence of PH1 has been reported to be 1ŌĆō3 per million populations in Europe,8,9 and PH1 accounts for 0.5ŌĆō2% of pediatric ESRD in Europe, North America, and Japan [10-12]. The prevalence of PH1 is higher in Mediterranean consanguineous populations, and the proportion of PH1 as a cause of pediatric ESRD is 10% in Kuwait [13] and 13% in Tunisia [14]. There have been no data on large cohorts except a few case reports of PH in Korea so far [6,7]. Although we did not analyze the prevalence of PH1, given that the patients in our study were limited to pediatric cases from three tertiary centers in Korea with a total population of approximately 50 million, the prevalence of PH1 in Korea might be similar to that in Western countries. In addition, PH1 accounts for 0.9% of pediatric ESRD according to the data of the chronic kidney disease registry of the Korean Society of Pediatric Nephrology (not published data). The marked male preponderance in our study is not explained.
In our study, 10 different AGXT mutations were detected, including a novel mutation, c.681-1G>A in intron 6. The GRHPR mutation (c.181G>A, p.Asp61Asn, rs371660673) detected in Patient 10 was predicted to be disease-causing by MutationTaster (http://www.mutationtaster.org/), and the minor allele frequency was reported to be 0.00005/6 on ExAC Browser (http://exac.broadinstitute.org/), 0.00008/1 on GO-ESP (http://evs.gs.washington.edu/EVS/), and 0.000008/1 on TOPMED (https://www.nhlbi.nih.gov/science/trans-omics-precision-medicine-topmed-program). The HOGA1 mutation (c.812G>A, p.Arg271His) detected in Patient 11 was also predicted to be diseasecausing by MutationTaster, and the minor allele frequency was reported to be 2/7524 in East Asians and 4/103800 in the total population on the ExAC Browser. However, a genetic diagnosis of Patients 10 and 11 was not confirmed because they only carried a single heterozygous mutation.
The most commonly detected AGXT mutation in our study was c.33dupC mutation with an allelic frequency of 44%. The run of eight cytosine residues (c.26_33) in exon 1 is known to be particularly prone to deletion/insertion mutations. Accordingly, c.33dupC mutation has been known as one of the three most common mutations across populations, p.Gly170Arg, c.33dupC, and p.Ile244Thr. However, the allele frequency of the c.33dupC mutation in our study was much higher than that in other studies (11ŌĆō15% of total AGXT mutant alleles) [15,16]. In addition, the p.Gly170Arg mutation, which is the most common mutation occurring at a frequency of approximately 30% [15,16] and shows responsiveness to pyridoxine [17,18], was not detected in our study.
Several studies have reported the possible association of homozygous c.33dupC mutation with poor prognosis, including earlier onset and more rapid progression to ESRD, in accordance with the complete lack of AGT protein [15,18-20]. However, there is a marked intra- and inter-familial phenotypic heterogeneity in families with homozygous c.33dupC mutation from ESRD in infancy to PH diagnosed in the second or third decade of life [21,22]. In our study, although the number of total patients was too small, patients with two truncating mutations including c.33dupC mutation, showed earlier ages of onset and more frequent retinal involvement than patients with one truncating mutation.
There has been no clear genotypeŌĆōphenotype correlation in PH1, except for pyridoxine responsiveness in some patients with PH1, particularly in those with homozygous p.Gly170Arg or Phe152Ile mutations [17,19]. Pyridoxine, vitamin B6, is a known cofactor of AGT and is effective in reducing urine oxalate excretion in about a third of PH1 patients [23]. Unfortunately, none of the patients in our study had p.Gly170Arg or p.Phe152Ile mutations.
Renal damage is inevitable in most patients with PH1 and ultimately progresses to ESRD. When the GFR drops to 30ŌĆō45 mL/min/1.73 m2, the kidney is unable to excrete the oxalate load effectively and oxalate is subsequently deposited in all tissues (systemic oxalosis) [24]. Since conventional hemodialysis and peritoneal dialysis do not eliminate sufficient amounts of oxalate, more intensive strategies such as short daily sessions of high-flux dialysis, nocturnal dialysis, or combinations of hemodialysis and nocturnal peritoneal dialysis, must be used to avert a continuous positive balance of oxalate and to limit systemic oxalosis [25]. For patients before reaching CKD stage 4, preemptive liver transplantation to avoid the complications of systemic oxalosis would be a logical approach [26]. However, this approach raises ethical issues given the risk of graft failure and death associated with the procedure [27]. Isolated kidney transplantation to correct ESRD in PH1 is frequently followed by recurrence of the disease in the graft kidney since the liver is the site of the metabolic defect. Therefore, combined liver and kidney transplantation is a reasonable choice for these patients, although kidney transplantation alone may be considered on an individual basis such as in adults with confirmed responsiveness to pyridoxine [1,10]. While dual transplantation is a reasonable choice for patients with CKD stage 4, sequential liver and kidney transplantation for patients with CKD stage 5 has the advantage that it provides sufficient time for the use of aggressive dialysis, which may mobilize some of the systemic oxalate burden and protect the kidney to be transplanted subsequently [1,28,29]. In our study, we adopted sequential liver and kidney transplantation for eight patients presenting with ESRD: three patients have already undergone this procedure and the rest are waiting for organ donation. According to the European pediatric registry [10], the 5-year survival rate for kidney transplantation alone and dual kidney and liver transplantation in children with PH was 14% and 76%, respectively
There is a limited experience with organ transplantation in patients with PH2. Although the ubiquitous tissue distribution of GRHPR favors kidney transplantation, some transplant recipients have undergone oxalate-related graft loss [5]. PH3 has not been associated with ESRD thus far, but there have been a couple of cases with impaired renal function [30].
In conclusion, the estimated prevalence of PH1 in Korea was similar to that reported in other studies from Europe, North America, and Japan. The c.33dupC was the most common AGXT mutation with an allelic frequency of 44%. The median age of onset was 3 months, and eight patients with PH1 already had ESRD at initial presentation. Although the number of total patients was too small, patients with two truncating mutations showed earlier ages of onset and more frequent retinal involvement than patients with one truncating mutation. Sequential liver and kidney transplantation was adopted for PH1 patients presenting with ESRD. A major limitation of this study was the small number of subjects. Therefore, a larger, nation-wide, multicenter study is needed.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print