Introduction
Renal impairment is a major complication of immunoglobulin A vasculitis (IgAV), and renal manifestations of IgAV are variable. IgA vasculitis nephritis (IgAVN) is a relatively benign disease in children [1]. However, long-term cohort studies have shown high sustained rates of severe proteinuria and renal dysfunction in these patients during adulthood, and patients with a history of IgAVN in childhood may experience a poor quality of life. Patients with IgAVN need lifelong care and observation. Trials have been performed to investigate early biomarkers and genes associated with poor prognosis to identify high-risk patients in whom IgAVN can progress to severe renal dysfunction and to establish an optimal treatment.
This systematic review of the recently published literature focused on the following questions:
a. Can biomarkers and genes predict renal involvement in IgAV?
b. If specific markers can identify high-risk patients in whom IgAVN can progress to severe renal dysfunction, can these predictors be used to prevent progression of renal disease?
c. Are there any new recommendations for IgAVN management?
Nomenclature used for immunoglobulin A vasculitis/Henoch-Sch├Čnlein purpura
The International Chapel Hill Consensus Conference (CHCC 2012) revised the nomenclature for vasculitis to include the pathophysiology of vasculitis in its name. Henoch-Sch├Čnlein purpura (HSP) is associated with IgA1-dominant immune deposits in small blood vessels (predominantly capillaries, venules, or arterioles). Therefore, the CHCC 2012 replaced the eponym ŌĆ£Henoch-Sch├Čnlein purpuraŌĆØ with IgA vasculitis to better define the pathophysiological features observed in this condition [2]. In this review, IgAVN has been used to replace the eponym Henoch-Sch├Čnlein purpura nephritis.
Diagnosis of immunoglobulin A vasculitis
In 1990, the American College of Rheumatology (ACR) proposed criteria to distinguish HSP (IgAV) from other vasculitides. Based on the ACR recommendations, the diagnostic criteria for HSP (sensitivity 87.1%, specificity 87.7%) include at least two of the following features: (1) age Ōēż20 years at disease onset, (2) palpable purpura, (3) acute abdominal pain, and (4) biopsy showing granulocytes in the walls of small arterioles/venules [3].
In 2010, the European League against Rheumatism and the Paediatric Rheumatology European Society (EULAR/PReS) jointly validated the criteria for HSP (IgAV) previously proposed by the ACR. These bodies suggested that IgAV should be diagnosed based on purpura or erythema (required criterion) predominantly observed over the lower extremities plus identification of one of these four features: (1) diffuse abdominal pain, (2) histopathological findings of typical leukocytoclastic vasculitis (LCV) with predominant IgA deposits or proliferative glomerulonephritis with predominant IgA deposits, (3) arthritis or arthralgia and, (4) renal involvement (proteinuria >0.3 g/24 hours or >30 mmol/mg of urine albumin to creatinine ratio in a spot urine morning sample, and/or hematuria : >5 red blood cells/high-power field or Ōēź2+ on dipstick or red blood cell casts in the urinary sediment) [4].
Compared with the ACR criteria, the EULAR/Paediatric Rheumatology International Trials Organisation (PRINTO)/PReS criteria focus on histopathological features of typical LCV with predominant IgA deposits or proliferative glomerulonephritis with predominant IgA deposits as useful criteria in all suspected cases with atypical distribution of purpura, and joint and renal involvement. Notably, the EULAR/PRINTO/PReS criteria showed better sensitivity and specificity than the ACR criteria [4].
Renal involvement in patients with immunoglobulin A vasculitis
Renal involvement occurs in 20ŌĆō50% of patients with IgAV [5,6]. Microscopic hematuria is the most common finding, and 25% of patients with IgAVN also show gross hematuria. Proteinuria occurs in 70% and isolated proteinuria in 9% of patients with IgAVN. Nephritis rarely precedes purpura onset, and nearly 97% of patients with nephritis develop urinary abnormalities within 6 months after the onset of other symptoms. Therefore, weekly urinalysis is warranted during the active phase of IgAV, followed by urinalysis performed each month for 6 months thereafter. Evidence of nephritis necessitates minimum 24-month follow-up despite resolution of urinary abnormalities [5-10].
Risk factors associated with renal involvement in patients with immunoglobulin A vasculitis
2. Genes and biomarkers
The human leukocyte antigen (HLA) gene complex and other genes associated with immunity and inflammation are known etiopathogenetic contributors to renal involvement in IgAV.
1) Genes associated with galactosylation of immunoglobulin A1
Aberrant galactosylation of IgA1 contributes to IgAV [13]. Reduced activity of the C1GALT1 (Gene ID: 56913) gene encoding core 1 ╬▓1,3-galactosyl transferase can cause aberrant glycosylation of IgA1 [14]. He et al. [15] reported that 1365 G/A allele frequency of the C1GALT1 gene was associated with increased susceptibility to IgAVN (P=0.0008, adjusted P'=0.004) with an odds ratio 1.716, 95% confidence interval 1.151ŌĆō2.560.
2) Nitric oxide synthase polymorphisms
Reportedly, cytokine-mediated-neutrophil-dependent injury to endothelial cells plays a key role in the pathogenesis of IgAV and may promote nitric oxide (NO) production [16]. Jiang et al. [17] investigated the potential association between nitric oxide synthase (iNOS) polymorphisms and the risk of progression of IgAV to IgAVN in a Chinese Han population. Children with IgAVN showed a significantly higher frequency of (CCTTT)12 repeats and A allele of rs2297518 than those with IgAV without nephritis.
3) Other biomarkers
The angiotensin-converting enzyme (ACE) gene [18], which is associated with vasomotor and endothelial function, the interleukin (IL)8 gene [19], which is associated with anti-inflammatory action, and the HLA-B35 (HLAb35) gene (reported by studies from Spain and the UK [20,21] were shown to be associated with IgAVN. Patients with HLA-B35 may be genetically more susceptible to recurrent episodes of IgAVN, precipitated by a heterogeneous group of infective stimuli resulting in a protracted illness with significant renal involvement. Reportedly, the ACE DD genotype or D allele is associated with IgAVN susceptibility in Asians [22].
3. Proteomic analysis
In 2015, He et al. [23] performed a comparative analysis of serum proteomes in IgAV (n=6), IgAVN (n=6), and controls (n=7) and identified a few differentially abundant proteins, such as C4a, serum amyloid A, angiotensinogen, and kininogen 1 in patients with IgAV and IgAVN. They specifically observed that angiotensinogen concentration was positively associated with IgAVN and could serve as a potential biomarker for progression of IgAV.
Long-term prognosis of immunoglobulin A vasculitis nephritis
The prognosis of IgAVN is better in children than in adults. A multicenter collaborative Italian study that included a mean 5-year follow-up reported that the risk of end-stage kidney disease (ESKD) was higher in adults than in children (15.8% vs. 7%) [1,28]. However, long-term prognosis in children with long life expectancy may yield different answers. The available literature describes 2 cohort studies that have included long-term follow-up (>20 years). A study in the UK investigated children with IgAVN who were treated in the early 70s and were followed-up until 1992 (median follow-up of 23.4 years) [29]. Notably, 82% of patients recovered among those with mild hematuria and proteinuria, and 44% showed persistent hypertension or kidney damage among those with nephritic and nephrotic syndrome; 18% of patients with early mild kidney disease showed persistent urinary abnormalities and 7.7% had ESKD. The overall ESKD incidence was 38.5% of all patients with IgAVN. Notably, severe clinical symptoms and significant renal biopsy findings were strongly associated with poor long-term prognosis. Regardless of the initial clinical symptoms, complications such as proteinuria and hypertension occurred in 16 of 44 pregnancies. A 24-year long-term cohort study in Finland investigated children with IgAVN treated between the 60s and 80s [30] and reported that 35% of patients with early severe renal symptoms showed persistent severe abnormalities. Mild urinary abnormalities persisted in 28% of children with mild renal symptoms and severe abnormalities in 11%. Overall, ESKD occurred in 10% of patients. The relative risk for poor renal outcomes was 5.0 in females and 2.0 in males. Initial renal biopsy findings were not associated with the risk of a poor renal outcome. Pregnancies were complicated by hypertension and/or proteinuria in 70% of women, and 56% of women with complicated pregnancies showed poor renal outcomes.
The aforementioned cohort studies indicated that pediatric IgAVN is no longer considered a mild disease. Proteinuria and hematuria may be associated with a 15% risk and a combination of nephritic-nephrotic syndrome may be associated with a 50% risk of progression to ESKD. These long-term studies highlight the importance of long-term follow-up in all patients during adulthood. Notably, all women with a history of IgAVN require close observation during and after pregnancy.
Prevention of progression to severe kidney disease and renal sequelae in patients with immunoglobulin A vasculitis nephritis
1. Can early treatment with corticosteroids or cytotoxic agents prevent progression of immunoglobulin A vasculitis nephritis to severe kidney disease?
The Kidney Disease: Improving Global Outcomes (KDIGO) group systematically reviewed the available literature [27] and reported that compared with no therapy or supportive therapy, early treatment with corticosteroids (CS) did not significantly contribute or contributed only minimally to severe kidney disease (nephrotic-range proteinuria, hypertension, or reduced kidney function). A long-term (20-year median follow-up) study in the USA reported that among patients who underwent a renal biopsy, 66% showed normal renal function and normal urinalysis, and 21% of patients progressed to ESKD. Cytotoxic agents served as the sole contributor to ESKD. However, this report did not describe any significant initial features that could help to identify children at risk of disease progression [31].
2. Can early aggressive treatment prevent exacerbation of renal disease based on identification of glomerular crescent deposition as predictors of renal sequelae?
A study investigating adults with IgAVN reported that greater crescent deposition was associated with more severe renal manifestations and a poorer treatment response; however, whether the percentage of crescent deposits is associated with a higher risk of late renal insufficiency remains controversial [32,33]. Multivariate analysis performed in a study by Pillebout et al. [34] showed that renal dysfunction and proteinuria at presentation, renal biopsy findings, the degree of interstitial fibrosis, percentage of sclerotic glomeruli, and glomerular fibrinoid necrosis were associated with poor renal prognosis. The percentage of crescent deposits is used as a criterion for severe nephritis warranting aggressive treatment in patients with IgAVN. The Cochrane collaboration concluded that there is a lack of evidence to support immunosuppressant administration in patients without severe nephritis considering that only a few poor quality trials have reported their use [27]. Niaudet et al. [35] suggested that early treatment might prevent progression of cellular crescent deposition to fibrosis and consequent chronic kidney disease. Plasmapheresis effectively removes IgA-containing complexes and prevents the development of and aids in resolution of crescents [36,37]. Further clinical trials are required to investigate more specific treatments to prevent monocyte influx into the BowmanŌĆÖs space. Selective blockade of IL-1 with IL-1 receptor antagonists and of tumor necrosis factor (TNF) with soluble TNF receptors markedly reduces crescent formation by reducing the expression of adhesion molecules and the recruitment of macrophages in experimental models of glomerulonephritis [38,39].
3. Can blood and urinary biomarkers, or genes detect the early signs of progression of immunoglobulin A vasculitis nephritis to severe renal dysfunction?
Institution of treatment only after renal biopsy leads to delayed management. Therefore, establishing less invasive and user-friendly laboratory tests are essential. Studies have investigated the role of genes, as well as blood and urinary biomarkers as predictors of progression of IgAVN to severe renal dysfunction.
1) Blood and urinary biomarkers
A French national cohort study (2017) [1] investigated blood and urinary biomarkers at the onset of skin rash and observed that children with IgAV with renal involvement at the time of diagnosis presented with serum galactose deficient (Gd)-IgA1 and urinary IgA, IgG, IgM, IL-6, IL-8, IL-10, and IgA-IgG and IgA-sCD89 complexes. Specifically, urinary IgA/cr and IgM/cr were significantly associated with IgAVN. Therefore, it is reasonable to conclude that patients showing these biomarkers are predisposed to subsequent renal dysfunction. However, a limitation of the study cannot be ignored; although 22 of the 33 patients (66.3%) showed severe disease with urinary protein creatinine ratio >1.0, the authors did not observe an association between histopathological findings and the aforementioned biomarkers.
2) Non-human leukocyte antigen region inflammatory and anti-inflammatory genes and vasomotor and endothelial function regulation genes
Studies have investigated the role of genes involved in autoimmune or vascular inflammatory pathways. A systematic review reported by Raquel et al.(2017) [40] reported that transforming growth factor beta 1, IL8, C-C Motif Chemokine Ligand 5, selectin P, angiotensin, ACE, paired box 2, endothelial NOS, vascular endothelial growth factor, and the Mediterranean fever gene were associated with vasculitis severity. Therefore, these parameters are useful indicators of renal injury and/or renal sequelae markers.
3) Long non-coding RNA
Long non-coding RNA (lncRNA) (characterized by >200 nucleotides and lacking coding potential) regulate coding genes on the level of >90% of all transcription and posttranscription [41,42]. They modulate small RNA and protein function and epigenetic modifications and serve as enhancer RNA. They display a highly tissue-specific pattern of expression and participate in several processes involved with development, health and disease. Pang et al. [43] reported that lncRNA were associated with the p53 signaling pathway and apoptosis-associated genes (AKT2 and FAS), as well as tumor protein 53, phosphatase and tensin homolog in patients with IgAVN. If further studies can conclusively prove the role of lncRNA in apoptosis, lncRNAs may be useful therapeutic targets in humans.
Treatment of immunoglobulin A vasculitis nephritis
The therapeutic recommendations for pediatric IgAVN currently followed in clinical practice are based on the KDIGO 2012 and SHARE 2019 guidelines. These guidelines are based on the recommendations of an expert panel and are not evidence based.
1. 2012 Kidney Disease: Improving Global Outcomes guidelines
These recommendations were similar to those established for the management of IgA nephropathy (IgAN) and suggest that the therapeutic protocol used to treat IgAVN in children should be similar to that used in adults [44]. However, the pathophysiological findings in IgAN differ from those in IgAVN. IgAVN in children tends to rapidly progress to a severe form and crescent deposition is more common in children than in adults with IgAVN [45,46]. Additionally, long-term follow-up in patients has shown that compared to the extent of crescent deposition, histopathological findings indicating chronic renal disease were more relevant as predictors of renal prognosis [33,34](Table 1).
These guidelines recommend the 3ŌĆō6 month administration of ACE inhibitors (ACEi) or angiotensin receptor blockers (ARBs) in children with proteinuria >0.5 g/day (including those with nephrotic syndrome) and glucocorticoid therapy is necessary in patients refractory to this treatment. Pediatric nephrologists are concerned that delaying the use of potentially effective therapy, such as methylprednisolone (MP) could predispose children to chronic renal disease in the long term [35,47,48].
2. 2018 Kidney Disease: Improving Global Outcomes glomerulonephritis guidelines update - Evidence summary in patients with immunoglobulin A nephropathy and immunoglobulin A vasculitis nephritis [49]
These guidelines were established in 2018 and state that compared to no therapy or supportive therapy, early treatment with CS contributed minimally or did not contribute at all to persistent kidney disease at 6 and 12 months and severe kidney disease in adults. Additionally, these guidelines state that the efficacy of cyclophosphamide (CYC) alone or mycophenolate mofetil (MMF) and azathioprine (AZA) is unclear in patients with severe kidney disease [49,50] (Table 2).
3. 2019 Kidney Disease: Improving Global Outcomes conference report
The KDIGO initiative organized a Controversies Conference on glomerular diseases in November 2017, which reviewed the role of steroids and immunosuppressive therapy in patients with IgAN but not IgA vasculitis [51,52]. The KDIGO guidelines suggest that CS (but not CYC/AZA) may reduce proteinuria, although the effect on renal function remains controversial [53].
4. 2019 European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-The SHARE initiative recommendation
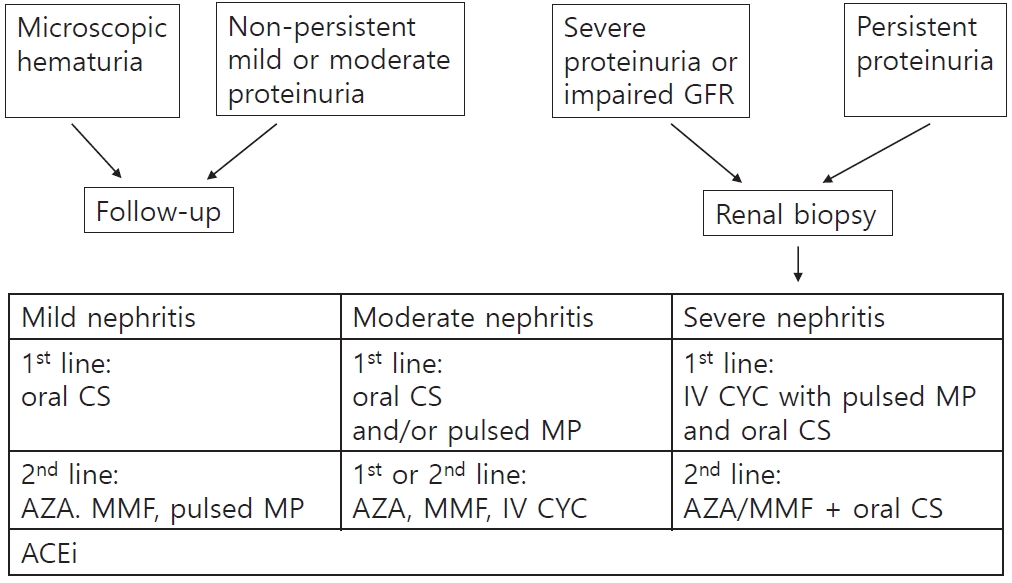
At the most recent SHARE consortium in 2018, based on consensus, 16 pediatric rheumatologists, nephrologists, and pathologists from seven countries developed recommendations for the diagnosis and treatment of IgAVN in children [54] (Fig. 1). Definitions to grade the severity of IgAVN and proteinuria are shown in Table 3. Recommendations for diagnosis of IgAVN are shown in Table 4. Similar to KDIGO guidelines, the SHARE initiative recommends the administration of CS, AZA, MMF, and CYC; however, this group concluded that calcineurin inhibitors or oral CYC is ineffective in these patients. Compared to KDIGO recommendations, SHARE recommends earlier administration of ACEi and early initiation of oral CS even in mild cases of IgAVN (Protin creatinine ratio PCR<100 mg/mmol=0.88 mg/mg). Oral prednisolone and/or pulsed MP is recommended as first-line treatment for moderate IgAVN (100<PCR<250 mg/mmol), <50% crescents on biopsy, renal dysfunction, usually International Study of Kidney Disease in Children histological class IIIb. AZA, MMF, or intravenous CYC may also be added to the first- or second-line regimen. Cyclosporin or oral CYC cannot be routinely recommended for moderate IgAVN. A renal biopsy is necessary in patients with IgAV showing severe proteinuria (>250 mg/mmol for at least 4 weeks, although a shorter duration of severe proteinuria also serves as a relative indication for biopsy) in those with persistent moderate (100 mg/mmol) proteinuria or impaired glomerular filtration rate). The treatment protocol for severe IgAVN (defined as >50% crescents on renal biopsy and renal dysfunction, ISKDC histopathological class IV-V disease, or nephrotic syndrome) is similar to that used for systemic small-vessel vasculitis with kidney involvement, e.g., antineutrophil cytoplasmic antibody-associated vasculitis. Treatment usually includes intravenous CYC with pulsed MP and/or oral prednisolone to induce remission, and lower doses of CS combined with AZA or MMF are administered as maintenance treatment.
5. Other recommendations for the management of immunoglobulin A vasculitis nephritis
Niaudet et al. [55] described the following therapeutic strategy for IgAVN in children: The authors are of the opinion that delaying the administration of potentially effective agents such as MP predisposes patients to chronic renal disease. On the other hand, patients with mild symptoms (i.e., microscopic hematuria, short-term macroscopic hematuria, or proteinuria <1 g/day) do not require specific therapy. Urinary protein excretion and serum creatinine is measured once per week for one month and every 2 weeks for 2 months thereafter to monitor disease progression. Children with more severe renal involvement (i.e., proteinuria >1 g/day, nephrotic syndrome, or crescentic glomerulonephritis on renal biopsy) are administered three doses of intravenous MP at a dose of 1 g/1.73 m2 (one dose daily, or on alternate days, for a total of three doses), followed by oral prednisone 30 mg/m2 once daily for one month and oral prednisone 30 mg/m2 every other day for 2 months.
6. Other treatment
1) Targeted-release budesonide formulation
Mucosal B lymphocytes located in PeyerŌĆÖs patches produce galactose deficient IgA1 (Gd-IgA1), which bind antiglycan IgG antibodies to form circulating immune complexes [56]. A recent genome-wide association study involving patients with IgAN identified susceptibility gene loci that are associated with intestinal mucosal immunity [57]. This pathogenesis suggests that local immunosuppression of mucosal B-lymphocyte activation and proliferation in PeyerŌĆÖs patches could attenuate Gd-IgA1 production. Targeted budesonide delivery to the distal ileum reduced proteinuria in patients with IgAN after 9-month treatment; a confirmatory phase 3 trial is currently underway [58,59].
More recently, Suzuki et al. [60] reported that renal biopsy specimens stained using a monoclonal antibody to Gd-IgA1 (KM55) showed positive results in patients with primary IgAN or IgAVN; however, tissues obtained from patients with hepatitis C virus (HCV) nephritis with IgA deposits did not show such positive staining. Overall, these findings suggest that the pathogenesis of HCV-nephritis with IgA deposits that is typically observed in some patients with IgAN differs from that observed in patients with primary IgAN; therefore, targeted budesonide administration produced the same results in those with IgAVN but not in those with secondary IgAN.
2) Eculizumab
Mesangial immune complex deposition triggers a series of downstream pathways, including complement activation via the mannose-binding lectin and other pathways, precipitating glomerular injury and tubulointerstitial scarring. Plasma C3a levels were significantly elevated in 35% of 46 adults with IgAN or IgAVN [61-63].
In this study, the plasma C3a level was significantly associated with serum creatinine concentrations but not with 24-hour urinary protein excretion.
Eculizumab, a humanized anti-C5 monoclonal antibody, has shown antiproteinuric effects in four patients with crescentic IgAN, and a phase 3 trial is currently underway [57].
3) Narsoplimab, a mannan-binding lectin-associated serine protease-2 complement inhibitor
Phase 3 trials are underway to study the renal indications for administration of the anti-mannan-binding lectin-associated serine protease (MASP)-2 monoclonal antibody (OMS721), which blocks the lectin pathway, and MASP-2 has received orphan drug designation for atypical hemolytic uremic syndrome and IgAN [64].
Mannan-binding lectin accumulates in the glomeruli in some patients and along with C4d deposition contributes to poor outcomes [65]. These data and the increasing understanding that clinical inhibition of the lectin pathway might show therapeutic benefit in these patients, suggest that the lectin pathway plays a key role in the pathogenesis of this condition.
5) Heparin
Coagulation abnormalities have been implicated in the pathophysiology of IgAVN. After review of the available literature, the KDIGO group reported the results of a study, which showed that aspirin and dipyridamole were ineffective in preventing persistent kidney disease [27]; however, another study has suggested that low-molecular-weight heparin injections administered over 8 weeks were associated with a lower risk of ESKD and improved proteinuria [68].
6) Plasmapheresis
Plasma exchange effectively removes circulating immune complexes during the acute phase of IgAVN. A study that investigated children with acute renal impairment, heavy proteinuria or both with histopathologically proven >grade 3 disease reported using early plasmapheresis alone in these patients. All children received at least nine exchanges with 70 mL/kg for 4.5% human albumin solution per session during the first 2 weeks and the last 20 mL/kg for fresh frozen plasma; 15 of the 16 children included in the study showed improvement after this therapy [37].
7) Targeting the angiotensin system
ACEi/ARB administration is recommended for the mild to severe proteinuria in patients with IgAN/IgAVN. Usually, the antiproteinuric effect of ACEi/ARB is attributable to their primary action on the intrinsic glomerular membrane where they enhance the barrier size-selective function in addition to efferent (postglomerular) arteriolar vasodilatation [69,70].
ACEi/ARBs inhibit mesangial cell (MC) stimulation that occurs after binding of angiotensin to specific MC receptors in patients with IgAN/IgAVN. Based on their mechanism of action, these drugs reduce proteinuria, MC proliferation, and renal fibrosis; however, endocapillary inflammation and crescent deposition are unaffected [71,72].
8) Antileukotriene agents
Montelukast (a leukotriene receptor antagonist) possesses potential anti-inflammatory properties, modulating the production of IL-6, tumor necrosis factor ╬▒ (TNF-╬▒), and monocyte chemotactic protein. Studies have reported that montelukast reduced systemic symptoms including purpura, abdominal pain, arthritis, proteinuria and hematuria in patients with IgAVN and also inhibited relapses during the first 3 months after treatment. However, this drug did not affect the outcome of nephritis at the end of follow-up [73-75].
9) Gluten-free diet
The mean serum GD-IgA1 level was significantly higher in systemic IgAV than in cutaneous IgAV. High GD-IgA1 levels may predict systemic IgAV [75]. Serum levels of Gd-IgA1-sCD89 complexes in patients with IgAN were associated with disease severity and progression but not with disease susceptibility [1,76]. Additionally, food components may contribute to the pathogenesis of IgAN. Gliadin (a component of gluten) is known to directly interact with sCD89, promoting IgAN development in a mouse model of IgAN [77]. Therefore, it is reasonable to conclude that a gluten-free diet may benefit patients with IgAVN as well [78].
10) Infection eradication
The onset of vasculitis is commonly preceded by an upper respiratory tract infection, which supports the role of a potential microbial etiopathogenesis in these patients. It is hypothesized that the antigenic structure of microorganisms resembles that of vessel walls in humans. Infection with these microorganisms could result in the production of cross-reactive anti-endothelial cell antibodies (AECA) known to be associated with vasculitis [79]. Interestingly, IgA AECA isolated from patients with IgAVN are known to bind to bovine glomerular endothelial cells, whereas serum from patients with IgAV did not show such binding [80] suggesting that antigen-specificity of AECA differs between patients with IgAV and IgAVN [81]. In 40 children with IgAV (6.7┬▒2.5 years), apical periodontitis, rhinosinusitis, and otitis media were identified in 53%, 48%, and 10% of the children, whereas among 11 children with IgAN (10.4┬▒2.5 years), these diseases were observed 55%,18%, and 0%, respectively. Administration of antibiotics and root canal therapy resulted in complete cure without onset of nephritis or recurrent attacks in 31 of 40 children with IgAV whereas in all of the IgAN patients with various histological grades achieved normalization of urinalysis by 7.2┬▒5.7 months after the start of steroid therapy, not by eradication of infectious foci [82].
11) Long non-coding RNA
Reportedly, lncRNAs could be potential biological markers for IgAVN and are being viewed as possible pathogenetic contributors to glomerular and tubular injury and interstitial nephropathy, suggesting that lncRNA could be useful future therapeutic targets in these cases [43].
Conclusions
A shared pathophysiological mechanism may underlie the presentation of IgAN and IgAVN. The 2012 KDIGO guidelines recommend that patients with IgAVN with severe proteinuria should receive the same treatment as that used for patients with IgAN. The progress of the disease differs between children and adults. Following the KDIGO recommendation in 2012, the 2019 SHARE group established guidelines for the treatment of IgAVN in children. A prominent difference between the recommendations of these bodies is that the 2019 SHARE group recommends steroid use even in cases of mild proteinuria, although the portion for immunosuppressants is similar. Future research on the pathophysiology of IgAN and clinical trials that are currently underway are expected to result in the development of novel drugs in patients with IgAVN.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print