Introduction
Growth and development in infancy can reflect infantsŌĆÖ physical health and be related to mental and social condition as well. The factors affecting infantile growth and development can be roughly divided into genetic and environmental, however, diverse medical or social conditions can affect infantile growth and development. Recently, national health screening for infants and children after 4 months of age has taken a place as a primary care service in Korea, and the early evaluation via the screening system has revealed an increased frequency of growth or developmental delay than was previously [1]. The medical approaches to growth or developmental delay necessarily include systematic wide spectra, including genetic, nutritional, endocrinologic, and neurologic aspects.
Diabetes insipidus (DI) is characterized by polyuria in spite of high serum osmolality, and is classified into central and nephrogenic according to the serum concentration of antidiuretic hormone (ADH). Decreased secretion of ADH from the pituitary gland is a pathogenesis of central DI, and decreased renal response to ADH is a pathogenesis of nephrogenic DI. Congenital nephrogenic DI is rare and is caused by mutations of arginine vasopressin receptor 2 (AVPR2) located on the X chromosome and aquaporin 2 (AQP2) on chromosome 12q13. The AVPR2 mutation accounts for 90% of congenital nephrogenic DI. The disease is involved more in males than in females [2]. Until now, 250 cases of mutations of AVPR2 have been reported in the world [3]. Congenital nephrogenic DI can be present in newborn babies within 2ŌĆō3 weeks of birth, but symptoms can be delayed in babies fed with breast milk [4]. Polyuria and polydipsia are characteristic symptoms of DI, and hyperthermia and mental retardation caused by repetitive dehydration can present in male. Sometimes poor gaining of weight, repetitive dehydration, or emesis can lead to misdiagnosis as gastroesophageal regurgitation [4]. Symptoms are often mild and hard to diagnose early in females [5].
Here we report a case of congenital nephrogenic DI with rare AVPR2 mutation, referred to our hospital because of developmental and growth retardations reported by the national health screening program for infants.
Case report
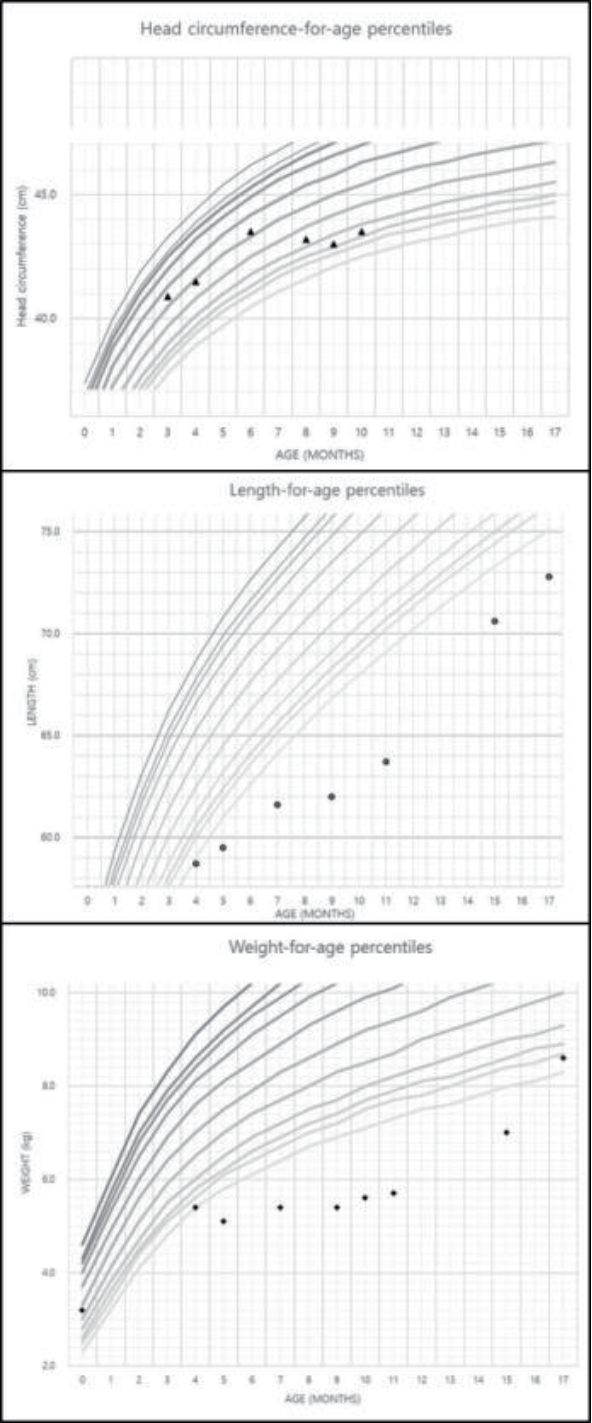
A male infant aged 5 months visited our hospital because of growth and developmental retardation reported by the national health screening program for infants. He was born via Cesarean section at 37 weeks of gestation after uneventful pregnancy and weighed 3.2 kg. Prenatal or perinatal problems were absent. The result of newborn screening test was normal. Family history was not specific. He had been vomiting 1ŌĆō2 times a day since birth and had been fed with 500ŌĆō700 mL of formula a day. Vital signs were stable, and anthropometric measurements at 5 months of age were as follows: height, 59.5 cm (0.1 percentile); weight, 5.1 kg (<0.1 percentile), and head circumference, 41.5 cm (19.1 percentile). We assessed the percentiles of anthropometric measurements using the 2017 Korean National Growth Charts for Infants, Pediatric Growth Standard (knhanes.cdc.go.kr). Skin turgor was intact, and dried lip was absent. Gross malformation was absent, but we observed hypotonic posture on physical examination. Auscultation of heart, lung, or abdomen was not specific. Overall developmental status was equivalent to about 3 months of age on the Korean developmental screening test (K-DST, www.mohw.go.kr). Although the patient did not present gross abnormality, we conducted routine blood tests with chromosomal analysis, methylation-specific PCR for Prader-Willi and Angelman syndrome, and genetic analysis of survival of motor neuron 1 (SMN1) for spinal muscular atrophy caused by repetitive emesis, poor weight gain, and hypotonia. However, routine blood tests and urinary analysis showed no specific and the performed genetic tests were either negative. Abdominal ultrasonography showed negative hypertrophic pyloric stenosis. Then we followed up the patient regularly together with rehabilitation therapy. Multidisciplinary care suggested a high-caloric formula, but he could not tolerate the formula, which aggravated his vomiting. He could roll from supine to prone position at 7 months of age, but head lagging persisted. The growth was still delayed as follows: height, 61.6 cm (<0.1 percentile); weight, 5.4 kg (<0.1 percentile); and head circumference, 43.5 cm (34.8 percentile).
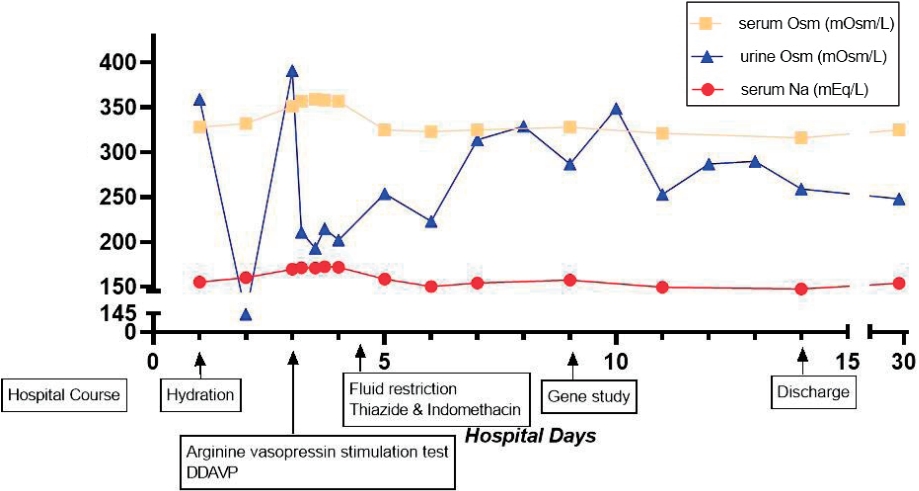
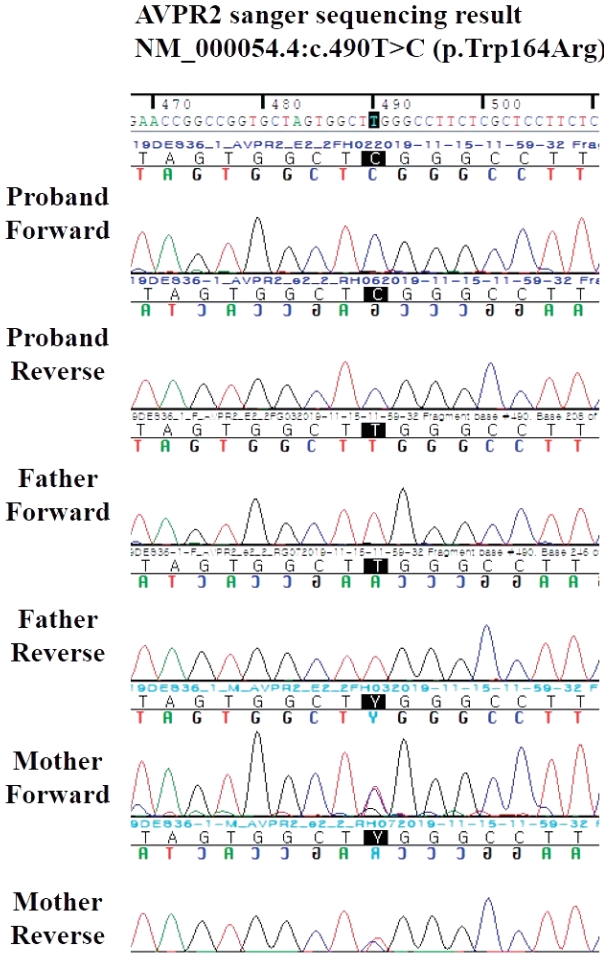
The patient visited again our hospital at 10 months of age because of fever. The vital signs were 115/74 mm Hg of blood pressure, 126 per minute pulse rate, 22 per minute respiration rate, and 38.2Ōäā body temperature. Anthropometric measurements were 68.8 cm in length (2.5 percentile), 5.2 kg in weight (< 0.1 percentile), and 45 cm in head circumference (37.5 percentile). He could roll back to stomach, but muscular hypotonia persisted. He was being fed 600ŌĆō700 mL of formula milk a day. Vomiting had persisted about twice a day since 5 months previously and it had not been aggravated recently. Diarrhea or decrease of urinary output was absent; so were respiratory symptoms like cough. Dried lip and depressed fontanelle were presented. Auscultations of chest and abdomen did not find abnormality. Laboratory tests were as follows: hemoglobin, 11.7 g/dL; white blood cell (WBC) count, 17,240/mm3; platelets, 187,000/mm3; total protein, 7.2 g/dL (reference range, 6.1ŌĆō7.9); albumin, 4.8 g/dL (1.9ŌĆō4.9); total calcium, 11.5 mg/dL (8.8ŌĆō10.8); phosphorus, 5.2 mg/dL (3.8ŌĆō6.5); blood urea nitrogen, 35 mg/dL (5ŌĆō18); creatinine, 0.4 mg/dL (0.03ŌĆō0.50); glucose, 80 mg/dL (50ŌĆō90); C-reactive protein, 4.1 mg/L (0ŌĆō5); sodium, 155.2 mmol/L (134ŌĆō144); potassium, 4.8 mmol/L (3.5ŌĆō6.1); and osmolality, 328 mOsm/kg (275ŌĆō295). Urinary WBC was absent, and urinary osmolality was 359 mOsm/kg (50ŌĆō1,400 according to water ingestion). Negative blood or urinary culture existed, but adenovirus was detected by respiratory viral real-time polymerase chain reaction. Fluid therapy and conservative care were initiated to manage hypernatremic dehydration and fever caused by the adenovirus. We serially checked serum sodium during rehydration therapy with 0.45% normal saline (150 mL/kg/day) over 48 hours. However, serum sodium steadily increased to 172.4 mmol/L. Serum and urinary osmolalities were 334 and 141 mOsm/kg, respectively. Urinary output was 199 mL/kg/day. We suspected diabetes insipidus and did a vasopressin stimulation test (0.33 U, subcutaneous route). However, serum sodium, serum osmolality, and urinary osmolality were not changed (Table 1). Fluid was not restricted during the stimulation test because of fear of shock caused by his weight and previous dehydration, which might contribute to an inadequate result. Therefore, we administered Desmopressin (Minirin┬«, 5 ╬╝g, Ferring International Center S.A., Saint-Prex, Switzerland) via nasal cavity; however, hypernatremia and low urinary osmolality persisted. (171.8 mmol/L, Table 1). We strongly suspected nephrogenic DI and initiated fluid restriction and diuretics on day 4 of admission. After fluid restriction (600 mL of formula milk a day), diuretics (hydrochlorothiazide, 1 mg/kg/day), and non-steroidal anti-inflammatory drug (dexibuprofen, 6 mg/kg/dose, bid) were initiated, serum sodium decreased to 150.4 mmol/L on day 14 of admission. (Fig. 1) ADH was over 80 pg/mL (normal range, Ōēż6.70) and radiologic studies, including renal ultrasonography and brain magnetic resonance, showed no specific finding. Genetic analysis was performed by a direct sequencing of AVPR2 and the c.490T >C (p.Trp164Arg) of missense mutation was detected. The mutation was also revealed in the mother, who, however, had no symptoms of nephrogenic DI (Fig. 2). He was discharged on day 15 of admission and has been checked regularly until now with taking diuretics.
Discussion
Congenital nephrogenic DI is a rare inherited disorder and is mostly caused by X-linked recessive AVPR2 genetic mutation. The AQP2 mutation leads to about 10% of nephrogenic DI with an autosomal recessive or dominant pattern. The epidemiology is not clear, but one study estimated about 8.8 of 1,000,000 male babies in Quebec, Canada [6]. Differences of mutations according to race is also unclear [2]. Until now, about 250 AVPR2 mutations among 300 families were reported in the world [2,3]. Most (50%) of them were missense mutation. Of them, p.Asp85Asn, p.Val88Asn, p.Arg113Trp, p.Arg137His, p.Ser167Leu, p.Arg171Cys, and p.Arg202Cys were often reported, and C to T or G to A base substitutions on hotspots of AVPR2 were causes of the mutations [3]. In our case, we detected a hemizygous c.490T>C (p.Trp164Arg) mutation on the X chromosome using Sanger sequencing of AVPR2; it was inherited from his mother. The variation was considered to be a disease-causing mutation, because there were characteristic symptoms and signs, such as hypernatremia and low concentrated urinary osmolarity under high AVP level in DI. The c.490T>C mutation is estimated to be highly related to the disease when considering in-silico prediction, distribution ratio among the population, and patient-specific symptoms, but there is only one report in the existing literature [2]. So currently, the mutation was considered as variant of uncertain significance according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines for interpretation sequence variant [7], however, it was considered as disease causing mutation in infant with typical symptom and sign of DI [8]. The mutation of hemizygous substitution of c.490T>C was previously reported once in Japan; however, to the best of our knowledge, this is a very rare mutation in the world and the first report of a rare X-linked recessive mutation in Korea [2].
X-linked recessive nephrogenic DI caused by AVPR2 mutation is very rare in females; however, diverse degrees of polyuria or polydipsia may be developed by skewed X-chromosomal inactivation [3]. The patient had a four-year-old sister. Even though we did not perform genetic analysis on her, and she was doing well, it should have been done, because, even though nephrogenic DI with AVPR2 mutation is mild in girls, it may develop later and be inherited by her future generations [5].
Growth and development are the major characteristics of infants and children. The status of growth and development during infancy can reflect mental and social conditions as well as health status. Individual infantile growth and development can be affected by many genetic and environmental factors, such as race, family, age, sex, chromosomal abnormality, nutrition, socioeconomic condition, and chronic diseases. Sometimes, caregivers cannot recognize an abnormality in the growth and development of their babies early. Recently, the national health screening program for infants and children after 4 months of age has taken a place as a primary care service in Korea. Hence the evaluation of infantile growth and development has been done much earlier, revealing a higher frequency of growth or developmental delay than had been known [1]. Although nephrogenic DI is rare, it should be considered as a possible cause in growth or developmentally delayed infants referred through the national health screening program for early diagnosis and treatment. In addition, since growth and development in infancy can affect health and social activity in childhood and adulthood, it is important to detect infants at risk earlier through the health screening program and to take diagnostic and therapeutic approaches [9].
At first, the patient was transferred due to retardation of development and growth on the National Health Screening Program for infants and children when he was 5 months of age. At that time, growth faltering and decreased muscle tone was checked, however, there was no specific finding on family or past medical history including feeding, urination and defecation. After 5 months since initial visit, fever caused by dehydration occurred and the authors diagnosed nephrogenic DI and began treatment. Although congenital nephrogenic DI is rare, early diagnosis and treatment are very important, because it usually leads to poor prognoses of seizure, growth and developmental retardation, or death caused by recurrent hypernatremic dehydrations [4,5,10-12]. In addition, since polyuria can lead to renal failure and bladder dysfunction caused by chronic dilation of the ureters and bladder, early diagnosis can affect renal prognosis [13]. Therefore, early diagnosis and appropriate treatment are required, but parenting person may not be sensitive to ingestion and excretion, and diagnosis may be delayed by history taking alone. Since patients with nephrogenic DI do not always have severe dehydration, pediatricians should suspect nephrogenic DI and perform serum and urine electrolytes and osmolarity if infants with poor weight gain are accompanied by recurrent episodes of fever [11].
Congenital nephrogenic DI is difficult to treat; the main therapeutic goal is adequate hydration with frequent water supply per oral or hypotonic saline (0.22%) [5,14]. Normal saline usually aggravates hypernatremia in congenital nephrogenic DI, except in case of hypovolemic shock. A low solute diet with low protein and sodium is recommended to decrease the excretion of urinary metabolite and sodium [14,15]. In this case, aggravation of vomiting after supply of a high-caloric formula (Infantrini®, Nutricia, Zoetermeer, Netherlands) might reflect a burden of hypernatremia or hyperosmolarity indirectly. He was fed with a commercially manufactured common formula milk and weaning food with frequent water supplementation. Thiazide was the first therapeutic approach in nephrogenic DI to increase urinary sodium concentration and osmolarity, and to decrease urinary volume by blocking the sodium-chloride cotransporter in distal convoluted tubule [14,15]. Besides, indomethacin can be considered as a therapeutic method in nephrogenic DI to increase urinary osmolarity and reduce urinary volume [15]. The mechanism of the prostaglandin synthesis inhibitor was unclear, but it can minimize urinary loss by decreasing the glomerular filtration rate [13] and increase resorption of water in collecting tubule by increasing the response to AVP [5]. Our case took hydrochlorothiazide and ibuprofen at first. However, he had been taking hydrochlorothiazide of 1 mg/kg/day because of fear of masking acute infection until the hypernatremia was under control. After that, he has been doing relatively well. He has grown to 8.6 kg in weight (2.3 percentile) and 72.8 cm in length (0.1 percentile), and can walk alone for a short time at 17 months of age (Fig. 3). Polydipsia had become apparent, and serum sodium is 140 mmol/L. Owing to a good compliance and tracing to the Korean national health screening program for infants and children, the patient might be diagnosed nephrogenic DI relative earlier than other previous cases [5,10-12] and prevent the progression of poor neurological or renal outcome.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print