Mechanism, clinical consequences, and management of dyslipidemia in children with nephrotic syndrome
Article information

Abstract
Dyslipidemia in nephrotic syndrome (NS) is often characterized by marked increases in the levels of total cholesterol, triglycerides, low-density lipoprotein cholesterol, and other lipoproteins, such as very low-density lipoprotein, intermediate-density lipoprotein, and lipoprotein(a). It has been suggested that impaired catabolism of lipoproteins and cholesterol is mainly due to decreased lipoprotein lipase and hepatic lipase activity, and increased biosynthesis of lipoproteins in the liver. The management strategies for dyslipidemia in patients with NS consist of lifestyle modification, lipid-lowering agents represented by statins, second-line agents such as fibrates and bile acid sequestrants, and lipid apheresis. Compared with dyslipidemia in adult NS patients, whose risks of atherosclerotic disease and progressive renal injury are considered high, clinical data on dyslipidemia in pediatric NS patients are limited. Therefore, it is necessary to pay more attention to the evaluation and management of dyslipidemia in pediatric patients with NS in clinical practice.
Introduction
Nephrotic syndrome (NS) is a clinical syndrome characterized by severe proteinuria, hypoalbuminemia, generalized edema, and dyslipidemia. It is mainly caused by the leakage of large amounts of protein from the blood into the urine due to malfunction of the glomerular filtration barrier. Primary (idiopathic) NS comprises >90% of cases of non-hereditary NS in children and adolescents, with pathological findings including minimal changes disease and focal segmental glomerulosclerosis. Although the incidence rate varies according to race and region, it is known to be approximately 2 per 100,000 children under the age of 15 to 18 years. In South Korea, the annual incidence of NS in pediatric patients is reported to be approximately 2 to 7 cases per 100,000 people, and the prevalence of NS is approximately 12 to 16 per 100,000 people [1,2].
It is well known that dyslipidemia in adult patients with NS significantly increases the risk of myocardial infarction and coronary artery disease, as well as progression of chronic kidney disease (CKD). However, studies on the risk of dyslipidemia in pediatric patients with NS are limited. Therefore, the importance of dyslipidemia management in pediatric patients with NS is often overlooked [3].
In this review, we describe the pathogenic mechanism, clinical consequences, and management of dyslipidemia in pediatric and adult patients with NS.
Pathogenesis of lipid abnormality
Lipids, mainly triglycerides (TG) and cholesterol, circulate in the body in the form of lipoproteins. Lipoproteins are formed from lipids packaged in apolipoproteins and phospholipids. The main forms of lipoproteins are chylomicrons, very low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), low-density lipoproteins (LDL), and high-density lipoproteins (HDL), which differ in composition and function.
It is known that there are three main pathways responsible for the production and transport of lipids within the body. First, in the exogenous pathway, dietary lipids ingested as food are packaged into chylomicrons in the intestinal mucosal cells and enter blood circulation through the lymphatic system. In the blood, TG, the main component of dietary lipids, is released as free fatty acids by lipoprotein lipase (LPL) in the capillary endothelium and transported to muscle, adipose tissue, and other peripheral tissues for absorption, and the remaining chylomicrons are transported to the liver for clearance. Second, in the endogenous pathway, VLDL generated in the liver is converted into IDL by LPL in the circulation and then absorbed into the liver by the LDL receptor (LDLR), releasing TG and free fatty acids in this process. In addition, IDL is transformed into LDL by hepatic lipase and LDL is removed from the blood by binding to LDLR in the liver and extrahepatic tissues. Third, the lipid metabolism pathway, known as reverse cholesterol transport, occurs via HDL. HDL is the so-called anti-atherogenic lipoprotein or good cholesterol because it captures cholesterol from peripheral tissues and other lipoproteins and transports them back to the liver [3].
Dyslipidemia in NS is characterized by an increase in total cholesterol (TC), TG, LDL cholesterol, and other lipoproteins, such as VLDL, IDL, and lipoprotein(a). In contrast, HDL remains almost normal, but the ratio of HDL cholesterol to TC is decreased [4]. This lipid abnormality is mainly caused by impaired catabolism of lipoproteins and cholesterol and, to a lesser extent, increased biosynthesis of lipoproteins in the liver.
It has been suggested that impaired lipoprotein clearance is due to decreased hepatic lipase and LPL activity in the endothelium and peripheral tissues such as muscle and adipose tissues [3,5,6]. In detail, as shown in Fig. 1., proprotein convertase subtilisin/kexin type 9 (PCSK9) regulates the expression of hepatic LDLR. In patients with NS, LDLR degradation increases as the intrahepatic expression of PCSK9 increases. Therefore, the uptake of LDL into hepatocytes is reduced and the clearance of LDL is disturbed [7-9]. In addition, when the ratio of free fatty acids to albumin in the blood increases due to proteinuria, circulating angiopoietin-like 4 levels increase, which leads to downregulation of hepatic lipase and a decrease in IDL clearance. Furthermore, as the permeability of the glomerular basement membrane increases and LPL activators decrease, LPL activity decreases and levels of IDL and VLDL increase [10,11]. Meanwhile, the level of immature HDL in the blood increases due to the reduction of cholesterol efflux through ATP-binding cassette subfamily A member 1, which is present in peripheral organs [3]. In patients with NS, the expression and activity of acetyl CoA acetyltransferase 2 (ACAT2) in the liver increases, which promotes cholesterol esterification and reduces the concentration of free cholesterol in cells. In an animal experiment using rats, it was reported that pharmacological inhibition of ACAT improved dyslipidemia and alleviated proteinuria [12,13]. Increase in 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase activity and cholesterol production have been observed in the liver of patients with NS [14,15].
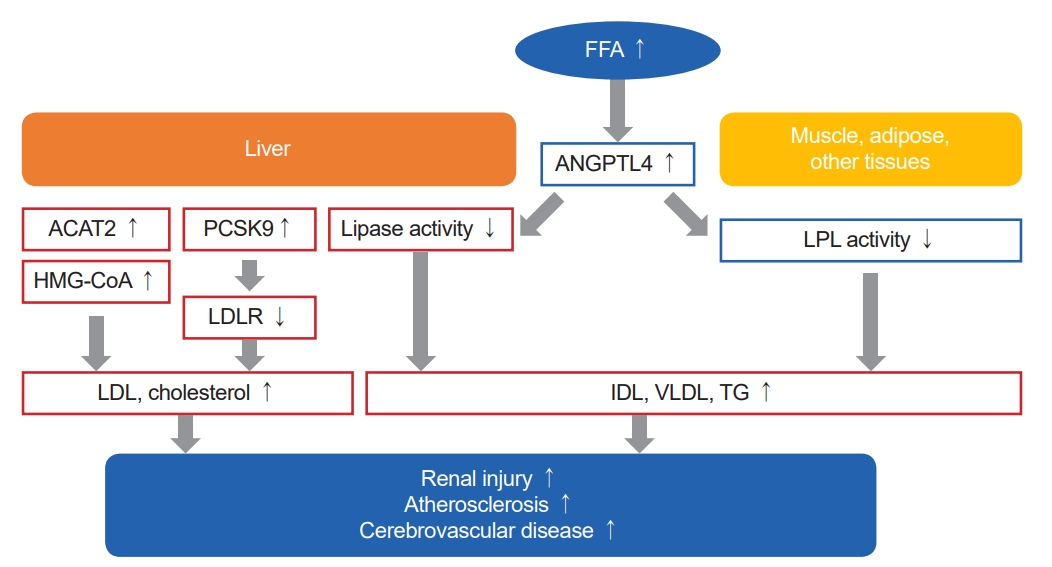
The pathophysiology and clinical consequences of dyslipidemia in nephrotic syndrome. In nephrotic syndrome, decreased hepatic lipase and lipoprotein lipase (LPL) activity in the extrahepatic tissues such as endothelium, muscle, and adipose tissue leads to impaired lipoprotein clearance, and thus increasing plasma levels of intermediate-density lipoproteins (IDL), very low-density lipoproteins (VLDL), triglycerides (TG). In addition, due to proteinuria and reduced free fatty acids (FFA) catabolism, as the ratio of FFA to albumin increases, levels of angiopoietin-like 4 (ANGPTL4) which inhibits lipase activity increase. Meanwhile, as the intrahepatic expression of proprotein convertase subtilisin/kexin type 9 (PCSK9) increases, low-density lipoproteins receptor (LDLR) degradation increases, resulting in reduced uptake of low-density lipoproteins (LDL) into hepatocytes. Furthermore, increased expression and activity of acetyl CoA acetyltransferase 2 (ACAT2) and 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase lead to elevation of plasma levels of LDL and cholesterol via increased synthesis and esterification of free cholesterol within the liver. Consequently, accumulation of oxidized LDL and IDL may cause glomerulosclerosis and other adverse effects on kidney by mesangial cell proliferation, podocyte injury, and tubular cell injury. Also, increased lipoproteins and cholesterol promote atherosclerosis represented by cerebrovascular disease.
Clinical consequences of dyslipidemia in NS
Atherosclerosis and cerebrovascular disease
In general, it is well known that dyslipidemia promotes arteriosclerosis and is a risk factor for myocardial infarction or cerebrovascular disease (CVD). Dyslipidemia exacerbates platelet hyperreactivity, which increases the risk of thrombosis, and is often accompanied by atherosclerosis. Although clinical studies to confirm the increased risk of CVD in patients with NS are scarce, a case-control study reported that non-diabetic adults with NS have a 5.5-fold significantly higher risk of MI (95% confidence interval, 1.6–18.3) compared to those without NS [16]. Recently, a case-control study comprising 66 children with NS and 128 age- and sex-matched controls showed that carotid-intima media thickness as a surrogate marker of CVD is significantly higher in children with NS aged over 4 years [17]. Therefore, in patients with persistent NS and accompanying dyslipidemia, especially in the presence of other cardiovascular risk factors, lowering cholesterol levels is important for preventing the progression of atherosclerotic lesions [18].
Renal injury
It has been suggested in several experiments and observations that dyslipidemia, accompanied with proteinuria and hypoalbuminemia, may increase renal organ damage and cause glomerulosclerosis by directly affecting mesangial cells, podocytes, and tubular cells, which is referred to as “nephrotoxicity hypothesis” [3,19].
Under normal conditions, LDL is metabolized and used by mesangial cells; however, when excess LDL is stored in the extracellular matrix in dyslipidemia, it is oxidized and causes an increase in cytotoxic agents such as prostaglandin E2 and tumor necrosis factor. These cytotoxic agents have the potential to damage the glomerular epithelial and endothelial cells, resulting in sclerosis.
In addition, it is hypothesized that increased free fatty acids bind to albumin and promote micropinocytosis of podocytes through lipid-binding G-protein coupled receptors, resulting in podocyte injury and loss, leading to end-stage renal disease [20,21]. Furthermore, it is reported that albumin-binding fatty acid may induce infiltration of macrophages and T lymphocytes into the tubulointerstitial space, leading to renal tubular cell injury and acute interstitial nephritis [22,23].
Several clinical studies, including prospective cohorts, reported that dyslipidemia could be a risk factor for renal injury represented as CKD progression. However, the findings were inconsistent among studies; some studies have reported that a high TC, TG, and LDL and a low HDL were associated with CKD progression, yet others did not [24].
Management of dyslipidemia in NS
The main approach for managing dyslipidemia is to treat the underlying renal disease that causes NS. Dyslipidemia usually improves when the underlying disease is treated with steroids, immunosuppressants, or angiotensin converting enzyme inhibitors/angiotensin-receptor blockers.
Lifestyle modification
A heart-healthy diet, physical activity, and weight reduction are recommended as the first-line treatments for pediatric patients with NS. In early studies, 20 NS patients with persistent proteinuria were administered a low-fat, low-protein, vegetarian soy diet rich in unsaturated fatty acids and fiber for 8 weeks instead of their usual diet. It was reported that lipid profiles (TC, LDL, HDL, apolipoprotein A, and apolipoprotein B), except for TG and proteinuria, were significantly improved due to the diet. However, lipid profiles and proteinuria returned to baseline levels after resumption of their original diet [25,26]. It was also suggested that omega-3 fatty acids decreased serum TG and postprandial chylomicron in patients with NS [27,28].
Pharmacological treatment: statins
Patients with NS should decide whether to initiate lipid-lowering medication based on CVD risk and renal function. Currently, there are no agreed upon guidelines for initiating lipid-lowering medications in patients with NS. Studies that provide clear evidence for the use of lipid-lowering medications are lacking. Therefore, it is necessary to consider the potential benefits and risks of such medication for each patient with NS.
Statins are the most commonly used drugs for the treatment of dyslipidemia in patients with NS. Statins inhibit HMG-CoA reductase competitively, reduce hepatic cholesterol production, and promote LDL absorption in blood. However, studies on statin administration in patients with NS are scarce. It was reported that TC and LDL were effectively reduced by 20% to 45% in adult NS patients treated with statins, but there was a lesser reduction in TG and apolipoprotein levels [29,32]. Meanwhile, in a meta-analysis including four randomized controlled trials (RCTs) using statins as lipid-lowering agents, only one RCT had a significant HDL improvement, and the others did not show a clear blood lipid improvement [33]. It was reported that statins also reduce lipoprotein(a) levels in NS patients in cases with high baseline values [34].
Studies on the treatment of dyslipidemia in pediatric patients with NS using lipid-lowering agents are very limited. Because long-term safety data are lacking and the U.S. Food and Drug Administration has approved its limited use in pediatric patients with familial hypercholesterolemia, the use of lipid-lowering agents is relatively low in pediatric patients compared to that in adults [3,35]. Most studies on pediatric patients with NS used statins, and it was shown that there is 30% to 40% lipid-lowering effect than before statin treatment [36,37]. Hepatotoxicity and muscle-related effects, including myopathy and rhabdomyolysis, have been reported as major side effects of statins, with common effects comprising gastrointestinal symptoms, such as diarrhea and musculoskeletal symptoms, as well as joint pain [35].
Pharmacological treatment: second-line agents
Fibrates, such as gemfibrozil, fenofibrate, and clofibrate, increase LPL activity, decrease TG production, and decrease plasma concentrations of TG and LDL. There have been small-scale RCT studies in NS patients that reported that gemfibrozil treatment reduced plasma TG concentrations by approximately 50% and the LDL concentration by 13% to 30% compared to placebo [38,39]. However, it is known that myopathy risk increases when fibrates are used in combination with statins [40].
Bile acid sequestrants such as cholestyramine and colestipol inhibit intestinal reabsorption of bile and block enterohepatic circulation of bile. Consequently, the expression of various liver enzymes involved in bile production increases, which in turn increases hepatic cholesterol breakdown and LDL absorption from the blood. In patients with NS, it has been reported that when cholestyramine was used, LDL was reduced by 19%, and when colestipol was used, it was reduced by approximately 30% [29,41]. However, the gastrointestinal side effects of bile acid sequestrants have been reported to be high; therefore, their use was often limited [3].
Nicotinic acid and ezetimibe can also be used to treat dyslipidemia; however, there are no available clinical data on patients with NS until now.
Monoclonal antibodies against PCSK9 (e.g., evolocumab and alirocumab) bind to and inactivate it, eventually increasing LDLR on hepatocyte surfaces to promote LDL uptake [3]. Recent studies reported that remission in NS patients was associated with a decrease in cholesterol and PCSK9 blood levels, and the LDL reduction effect in the group treated with a PCSK9 inhibitor was significant [42-44]. In addition, ACAT inhibitors were reported to improve proteinuria and dyslipidemia in NS animal models [13].
Lipid apheresis
Lipid apheresis has been used to treat patients with homozygous familial hypercholesterolemia and has recently been applied to the treatment of dyslipidemia in NS patients [3]. It has been reported that adult and pediatric patients with steroid-resistant NS treated with lipid apheresis with or without steroid showed reduced proteinuria and improved lipid profiles. This effect might be explained by the improvement in dyslipidemia, removal of autoantibodies, reduced potential vascular permeability factors and inflammatory cytokines, and improved responsiveness to immunosuppressants [20,45-47].
Conclusions
In this article, the authors reviewed recently described mechanisms, clinical impacts, and several treatment methods for dyslipidemia in patients with NS. Compared with adult patients with NS whose risks of atherosclerotic cardiovascular disease, such as myocardial infarction or coronary arterial disease, are high, studies on dyslipidemia in pediatric patients with NS are still lacking. However, there are also possible risks of atherosclerotic cardiovascular disease and progressive renal injury due to severe dyslipidemia, even in pediatric NS patients. In conclusion, more attention should be paid to the screening and treatment of dyslipidemia in pediatric patients with NS in clinical practice.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
None.
Author contributions
All the work was done by HSB.