Introduction
Kleefstra syndrome (KS) is a rare genetic disease characterized by distinctive facial features, intellectual disability, congenital heart defects, epilepsy, recurrent infections, and renal abnormalities. Vesicoureteral reflux (VUR) and renal cysts are the primary renal complications, which may be accompanied by chronic renal insufficiency and severe constipation in addition to hydronephrosis [1,2]. Herein, we present a case of KS in a patient born with ambiguous genitalia and an imperforated anus after being diagnosed with VUR and rectourethral fistula. This case report highlights the importance of a multidisciplinary approach to manage the frequent structural and functional problems in patients with anorectal or genitourinary anomalies and associated recurrent urinary tract infections (UTI).
Case report
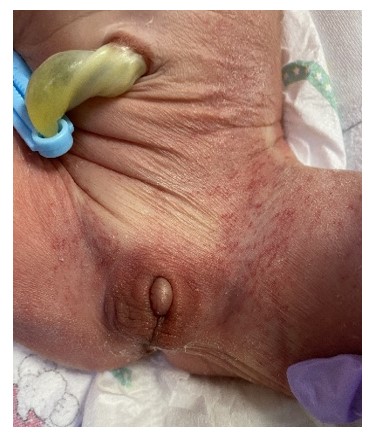
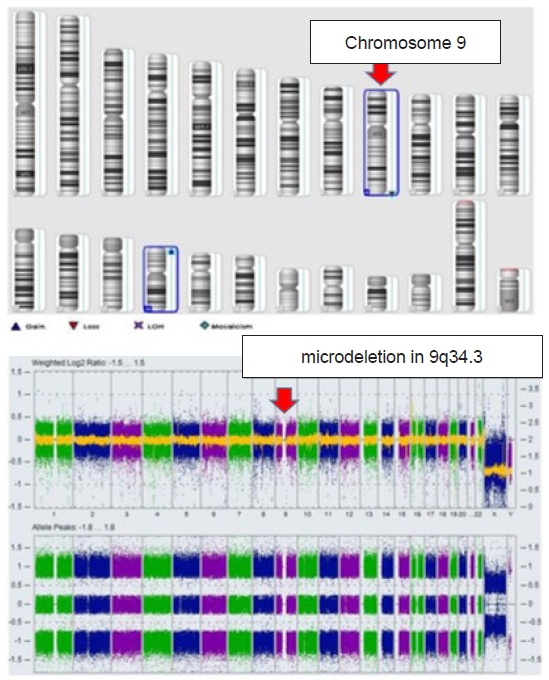
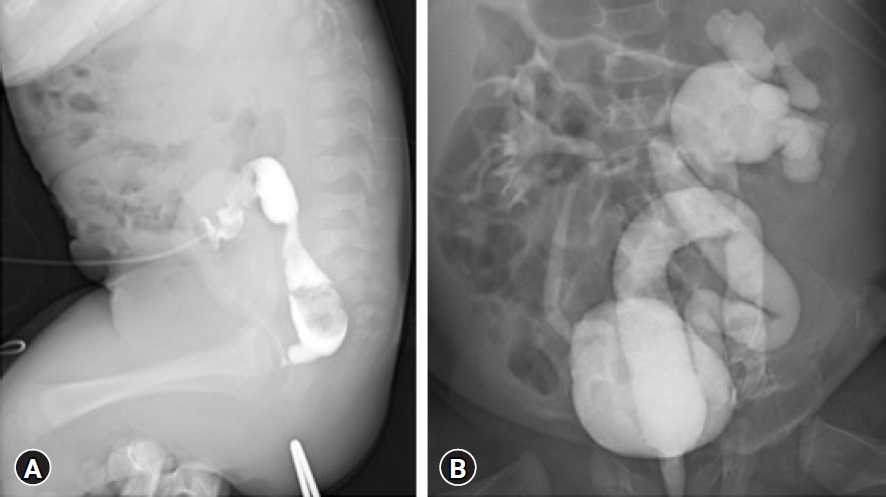
A 1-day-old boy with ambiguous genitalia (Fig. 1) and an imperforated anus was transferred to the neonatal intensive care unit (NICU) of our hospital. He was born at 38 weeks and 5 days of gestation by normal spontaneous vaginal delivery, before which he showed no special findings on prenatal examinations. At birth, his weight was 2,870 g (14 percentile), height 50 cm (51 percentile), and head circumference 31 cm (1 percentile) compatible with microcephaly. He had no specific family history; however, physical examination showed facial hypertelorism, arched eyebrows, midface hypoplasia, short nose with upturned nares, protruding tongue with everted lower lip, and downturned corners of the mouth. On the 3rd day of hospitalization, colostomy was performed by a pediatric surgeon. Genetic studies were further performed for multiple anomalies, including dysmorphic face, micropenis, and imperforated anus. The karyotype (46, XY) was nonspecific; however, the microarray test showed a microdeletion in 9q34.3, which is compatible with KS (Fig. 2). The patient experienced urosepsis on the 12th day of life. At that time, Enterococcus faecium was identified in his urine and blood cultures, and he was treated with antibiotics. Renal ultrasonography for UTI examination showed severe hydronephrosis (grade 4, based on the grading system of the society for fetal urology) in the left kidney, while 99mTc-dimercaptosuccinic acid renal scan revealed diffuse decreased uptake and atrophic changes in the left kidney. Approximately 3 weeks after the first episode of UTI, a second episode occurred, and antibiotic prophylaxis was initiated. On loopogram examination performed on the 53rd day after birth, high or intermediate-type imperforated anus with rectourethral fistula was confirmed (Fig. 3A). Despite antibiotic prophylaxis, the third episode of UTI occurred 40 days after the second episode. Voiding cystourethrography revealed bilateral VUR with grade 2 on right, grade 5 on left, and rectourethral fistula (Fig. 3B). The patient was discharged from the NICU while continuing antibiotic prophylaxis at 3 months of age. However, he was subsequently readmitted to our hospital because of a fourth episode of UTI on the 102nd day after birth.
At approximately 6 months of age, he was admitted to the department of pediatric surgery, and anorectoplasty combined with repair of rectourethral fistula was performed. Three months later, the patient underwent colostomy repair to complete the correction of the imperforate anus. Despite the completion of surgical treatment for anorectal anomalies, episodes of UTI recurred several times. To discontinue these recurrent episodes of UTI, we referred to the urology department for surgery for VUR. The pediatric urologist replied that the patient was likely to develop several critical complications of open surgery for VUR due to the previous operation. So endoscopic Dexol injection was performed on the left VUR at around 11 months of age, in accordance with the opinion of the pediatric urologist. However, despite the endoscopic Dexol injection, his UTI recurred, and high-grade VUR in the left kidney was maintained. On consultation with a pediatric urologist, we thought that increased bladder pressure associated with constipation and the initiation of defecation through the anus was presumed to be another possible cause of UTI. Therefore, we conducted a urodynamic study and confirmed hyperactive detrusor and external sphincter findings. We decided to administer anticholinergic drugs and begin clean intermittent catheterization (CIC) three to four times a day. Thus far, he is continuing CIC and there has been no recurrence of UTI during regular follow-ups with this management in spite of the existence of high-grade VUR.
Discussion
KS is a rare genetic disease with an autosomal dominant inheritance [1]. The major phenotypic characteristics result from defects in the euchromatic histone lysine methyltransferase 1 (EHMT1) gene, which is caused by a point mutation or microdeletion in chromosome region 9q34.3 [1]. Most cases have been reported to be de novo; however, cases of familial pathogenesis have also been reported. Although the prevalence of KS is unknown, genome-wide studies have indicated that it accounts for approximately 0.2% of neurodevelopmental disorders. Patients with KS have characteristic facial features, including a flattened area at the back of the skull, small head, underdevelopment of the midface, fusion of the eyebrows above the bridge of the nose, cupid bowed upper lip, fully everted lower lip, protruding tongue, and lower jaw. These patients are also characterized by moderate-to-severe intellectual disabilities with delayed speech development. Other features observed in KS include congenital heart defects, epilepsy, recurrent infections, and hearing problems. Male patients may also have genital problems, such as hypospadias, cryptorchidism, micropenis, and kidney defects with VUR, hydronephrosis, kidney cysts, and chronic kidney disease [2]. KS is diagnosed based on distinctive clinical features and molecular genetic testing, such as chromosomal microarray, intellectual disability gene panels, whole exome, whole genome sequencing, or targeted genetic testing. Management of KS involving special education, and speech, physical, occupational, and sensory integration therapies is recommended from an early age. Because cardiac, renal, and intestinal complications may develop, regular check-ups are recommended and the patients should be followed up for life. Although the prognosis of KS is generally favorable with low fatality, it varies depending on the presence or severity of comorbidities.
Most cases of KS thus far reported in the Republic of Korea have been diagnosed as neurological developmental disorders [3-5]. According to a study evaluating the benefits and yield of array comparative genomic hybridization, two out of 87 pediatric patients with neurologic disease who had developmental delay, dysmorphic face with features such as hypertelorism, a short nose, and down turned corners of the mouth (a phenotype consistent with KS) had a deletion of the EHMT1 gene [3]. In addition, in a previous report, among 40 patients with microcephaly, one patient with primary microcephaly, epilepsy, and congenital heart disease showed a deletion at 9q34.3 [4]. In a study assessing the diagnostic utility of chromosomal microarray testing, one out of 649 patients with developmental or intellectual delay had a 320-kb 9q34.3 sub-telomeric deletion, and was diagnosed with KS [5]. Therefore, this is the first reported Korean case of genitourinary and anorectal anomalies in KS.
According to a retrospective review of anorectal malformations, comorbidity of genitourinary anomalies was high in children with anorectal malformations, among whom VUR was reported to be high [6]. van der Steeg et al. [7] also reported that congenital anomalies of other organs were present in 89% of patients with anorectal malformation of the rectovesical fistula type, among whom renal anomaly was the second most common anomaly after sacral anomaly. In this retrospective Dutch cohort study, various anorectal complications, including bowel problems, UTI, and urinary retention, were reported to develop after reconstructive surgery, and most patients required bowel management [7]. Finally, regarding urological and renal outcomes during 5-year follow-up, recurrent UTI occurred in 38% of patients, and it was confirmed that approximately 25% of patients required CIC, as in our patient [7]. Likewise, in patients with anorectal anomalies, accompanying genitourinary malformations, including VUR, appear with a high probability, resulting in complications and poor outcomes. Therefore, early evaluation and treatment of accompanying malformations of the genitourinary system are necessary in patients with anorectal malformations. Also, any physician treating a patient with anorectal malformation should consider screening for genitourinary abnormality.
This is the first reported case of KS in the Republic of Korea in which anorectal and genitourinary malformations were resolved by step-by-step surgery, emphasizing the need for early detection and diagnosis of KS and evaluation of their comorbidities.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print