Navigating the landscape of clinical genetic testing: insights and challenges in rare disease diagnostics
Article information

Abstract
With the rapid evolution of diagnostic tools, particularly next-generation sequencing, the identification of genetic diseases, predominantly those with pediatric-onset, has significantly advanced. However, this progress presents challenges that span from selecting appropriate tests to the final interpretation of results. This review examines various genetic testing methodologies, each with specific indications and characteristics, emphasizing the importance of selecting the appropriate genetic test in clinical practice, taking into account factors like detection range, cost, turnaround time, and specificity of the clinical diagnosis. Interpretation of variants has become more challenging, often requiring further validation and significant resource allocation. Laboratories primarily classify variants based on the American College of Medical Genetics and Genomics and the Association for Clinical Genomic Science guidelines, however, this process has limitations. This review underscores the critical role of clinicians in matching patient phenotypes with reported genes/variants and considering additional factors such as variable expressivity, disease pleiotropy, and incomplete penetrance. These considerations should be aligned with specific gene-disease characteristics and segregation results based on an extended pedigree. In conclusion, this review aims to enhance understanding of the complexities of clinical genetic testing, advocating for a multidisciplinary approach to ensure accurate diagnosis and effective management of rare genetic diseases.
Introduction
The landscape of diagnostic methodologies for genetic diseases has undergone a remarkable transformation, leading to the discovery of thousands of genetic conditions. Currently, around 7,000 rare diseases have been identified, with an estimated 80% attributed to genetic factors, and 50% manifesting during childhood [1-4]. Early genetic diagnosis has proven particularly beneficial for pediatric patients, offering significant cost savings and enabling long-term disease management [5]. Recent technological advancements and cost reductions have made various genetic tests integral to diagnostic evaluations in clinical practice. Next-generation sequencing (NGS) stands out due to its ability to simultaneously identify variants across multiple genes. This capability has led to cost efficiency and high diagnostic rates, especially for diseases characterized by genetic heterogeneity. Nevertheless, the challenge of interpreting the vast array of variants generated by NGS is non-trivial and often requires additional resources for validation. NGS may not be the optimal choice for single-gene diseases distinguishable by characteristic clinical features, and it is not considered the gold standard for detecting certain genetic variations such as copy number variations (CNVs) or short tandem repeats (STRs). Therefore, a critical aspect of the diagnostic process is accurate clinical assessment, followed by the judicious choice of testing methodology and careful interpretation of test reports by clinicians.
This review is divided into two sections. The first section covers various clinical genetic testing methodologies, discussing the types of tests and factors influencing test selection. The second section focuses on the interpretation of germline sequence variants, a common outcome of NGS. This structure allows for a comprehensive exploration of clinical genetic testing, ranging from broad methodologies to the detailed interpretation of specific variants.
Comprehensive review of clinical genetic testing in rare genetic diseases
Overview of genetic testing modalities
Clinical genetic testing has become a pivotal component in diagnosing rare diseases, offering a broad range of tests available in clinical settings. Each genetic test is performed based on specific principles and has corresponding indications. There are three primary types of genetic testing: cytogenetic (chromosomal), DNA (molecular), and biochemical. Chromosomes, thread-like structures made up of DNA can be observed under a microscope after specific staining since the 1980s [6]. Microarray techniques, designed to identify small-unbalanced rearrangements, had developed and now feature diverse established platforms [7-9]. This method is recognized as a first-tier cytogenetic test for patients with developmental delay/intellectual disabilities, multiple congenital anomalies, and autism spectrum disorder [10-13]. Fluorescence in situ hybridization provides a unique advantage by visually mapping genetic material within a cell, facilitating the identification of structural chromosomal abnormalities [14,15]. Molecular genetic testing, the most frequently performed category, assesses single DNA loci, single genes, or multiple genes. Sanger sequencing, a traditional method and the gold standard for identifying single nucleotide variations is renowned for its high accuracy in analyzing short DNA sequences [16]. The polymerase chain reaction (PCR), a versatile tool widely used to amplify small DNA segments, is essential in various genetic tests, including those for infectious diseases [17-19]. Multiplex ligation-dependent probe amplification (MLPA), a robust method for detecting deletions and duplications of up to 50 nucleic acid sequences, proves invaluable for diagnosing various genetic disorders [20,21]. Some biochemical genetic tests, such as Southern blotting, while less common, still play a role in identifying specific DNA sequences in larger DNA samples [22,23]. NGS, a revolutionary form of molecular genetic testing, enables the rapid sequencing of large stretches of DNA or RNA, dramatically transforming the fields of genomics and molecular biology and facilitating a broad range of applications [24-27].
Critical factors in genetic test selection
In clinical practice, each genetic testing method is tailored to a specific indication. In terms of the detection range, some tests focus on single loci, while others, like NGS, can cover the entire genome. However, NGS is not the gold standard for detecting CNVs or STRs. Despite a decrease in cost and processing time, NGS remains more expensive and has a longer turnaround time (TAT) compared to traditional tests. Clinicians must take these factors into account when selecting tests (Table 1). An accurate and specific clinical diagnosis is crucial, as it guides the identification of potential causative genes and common types of genetic variation. For example, in cases clinically diagnosed with Fragile X syndrome, the first-tier confirmatory tests are Southern blotting or PCR targeting FMR1. Similarly, for pathologically confirmed Alport syndrome with a family history of X-linked inheritance, testing for the COL4A5 gene using sequencing and MLPA is advisable, as 10% to 15% of these cases involve exon-level deletions or duplications [28,29]. A precise clinical diagnosis facilitates targeted testing, thereby reducing both the length of the diagnostic journey and associated costs. Although essential, a detailed clinical assessment, including examination findings, routine laboratory tests, and specific biomarkers such as pathological findings, is not extensively discussed in this article.

Comparative characteristics of different genetic testing methodologies
The urgency of diagnosis is vital in certain cases, necessitating consideration of clinical severity and TAT. Many inherited metabolic disorders can lead to irreversible damage if not promptly managed [30]. In cases of serious and rapidly progressive illnesses, quick decision-making is essential. Some patients may find themselves in a situation where they have a serious and rapidly progressive illness, requiring swift decision-making. In such cases, opting for tests with the fastest available results rather than the most cost-effective sequence of tests may be clinically justified. Rapid genomic sequencing, which significantly shortens TAT, is increasingly used for patients with suspected medically actionable disorders or those in intensive care units [31-33].
Finally, cost-effectiveness is a critical factor in healthcare, particularly for the diagnosis of rare diseases. The introduction of advanced genomic technologies like NGS has broadened our diagnostic scope, but their higher initial costs necessitate careful test selection. Cost-effectiveness typically involves starting with less expensive tests, followed by more comprehensive and sensitive yet costlier methods if the initial results are inconclusive. This tiered approach balances the need for thorough genetic analysis with budget constraints and enhances patient care by providing efficient and economically viable genetic testing strategies. Economic considerations in genetic testing go beyond cost reduction; they focus on maximizing the value that each test brings in terms of clinical outcomes and informed healthcare decisions. Although recent studies have demonstrated the cost-effectiveness of genome sequencing as a primary diagnostic tool in certain rare disease groups [34-36], the heterogeneity in study inclusions and cost-effectiveness parameters cast doubt on the generalizability of these results, highlighting the need for further well-designed studies. Additionally, costs and availability differ by country; for example, in Korea, the current insurance system officially covers only limited multi-gene panel tests, necessitating a different approach to the diagnostic strategy.
Deciphering genetic variants: analysis and clinical correlation
Various genetic variants contribute to the development of rare genetic disorders. However, this section focuses specifically on germline sequence variants, which have become increasingly significant due to the rise in NGS utilization. This technology generates numerous variants of uncertain clinical significance. The process of identifying variants through NGS data involves the following sequential steps: variant calling, annotation, and the evaluation of disease causality. In this section, we will delve into the process of evaluating the final disease causality of a variant as reported in the test, particularly from the clinician’s perspective.
Germline variant classification
Classifying germline sequence variants is a crucial step in genetic testing, guided by comprehensive criteria established by leading organizations such as the American College of Medical Genetics and Genomics (ACMG) and the Association for Clinical Genomic Science (ACGS). The widely recognized and utilized ACMG guideline categorizes variants into five groups: pathogenic, likely pathogenic, uncertain significance, likely benign, and benign. This classification relies on various factors, including the population database, gene characteristics, prior reports, segregation results, computational predictions, and functional studies [37]. ACGS provides a framework aligned with ACMG, underscoring the significance of clinical context and multidisciplinary expert consensus in variant interpretation [38]. Typically, laboratory physicians report pathogenic and likely pathogenic variants and occasionally variants of uncertain significance based on their policies. Despite global application, these guidelines are not without limitations. Variability in interpretation among clinicians and laboratories may result in inconsistent variant classification [39]. The databases crucial for variant interpretation are still evolving and may not sufficiently cover population-specific variations. Furthermore, these guidelines often prioritize molecular characteristics over the complete clinical profile of the patient, potentially leading to less personalized assessments. There is a growing demand for clearer variant interpretation, prompting efforts to modify and update these guidelines to better suit specific genetic or clinical subgroups [40-44]. However, studies based on large-scale cohorts have yet to provide universally applicable criteria for variant interpretation. Consequently, the final interpretation of variants, particularly those of uncertain significance, remains challenging when relying solely on guidelines. Achieving accurate interpretation and application of these findings necessitates a comprehensive, patient-centric approach by clinicians [45-48].
Clinical correlation and personalized interpretation of variants
Before finalizing the results for a patient, clinicians must consider various factors in alignment with the reported genes/variants. The first factor is key phenotypes which include the age of onset, primary clinical symptoms, and disease progression. While theoretically, patients with the same variant may exhibit similar clinical symptoms, it is common to find individuals with the same variant presenting a wide range of diverse phenotypes, even within the same family (Fig. 1). This necessitates careful consideration [49]. For instance, the autosomal dominant polycystic kidney disease, is well known for various phenotypes, ranging from simple cyst to early end-stage renal disease, illustrating variable expressivity (Fig. 1A) [50]. The GLB1 gene, known to cause GM1-gangliosidosis with symptoms including progressive cerebral degeneration and developmental regression, is also responsible for Morquio disease. Morquio disease is characterized by multiple skeletal abnormalities and coarse facial features without clear neurological symptoms, exemplifying phenotypic variation or pleiotropy (Fig. 1B) [51,52]. A detailed family history assessment is crucial for patients with genetic disorders. The inheritance patterns of the identified genes should align with the family history and segregation results. If a gene associated with an autosomal dominant Mendelian disorder is documented, the variant should not be present in the asymptomatic parents, indicating de novo variation. Family test results, reflecting inheritance patterns, are critical and are incorporated into the ACMG and ACGS guidelines [37,38]. Unmatched results necessitate a reevaluation of the diagnosis. Notably, some diseases exhibit incomplete or reduced penetrance, explaining why asymptomatic family members carry the causative variant (Fig. 1D) [53,54]. Factors influencing penetrance include variant types, gene expression levels, epigenetic changes, gene-environment interactions, and genetic modifiers [55]. However, these theoretical factors often provide limited practical insight in clinical settings. Clinicians primarily rely on previous clinical reports and databases for practical information. For example, consider the case of the COL1A1 gene, associated with osteogenesis imperfecta, a rare connective tissue disorder. Osteogenesis imperfecta is known for its variable expression and incomplete penetrance, as documented in several clinical studies [56,57]. When a variant of the COL1A1 is identified in a family, and an asymptomatic family member carries the variant, it can be confirmed as causative if the phenotype matches the disease and co-segregation results for the rest of the family members (ideally from an extended pedigree) align with the known inheritance pattern, given the recognized incomplete penetrance of the gene. Similarly, specific diseases or gene subgroups, such as inherited retinal disease, hereditary spastic paraplegia, polycystic kidney disease, and renal agenesis/hypoplasia, are known to exhibit incomplete penetrance [58-61]. In summary, clinicians can ascertain the final causality of a variant using case-level clinical correlations. This determination hinges on a comprehensive assessment in which all pieces of the puzzle, including phenotypic consistency with the gene, family test results based on an extended pedigree, and research findings from existing databases, fit together harmoniously. If any of these factors are missing or inconclusive, a conservative interpretation approach should be adopted.
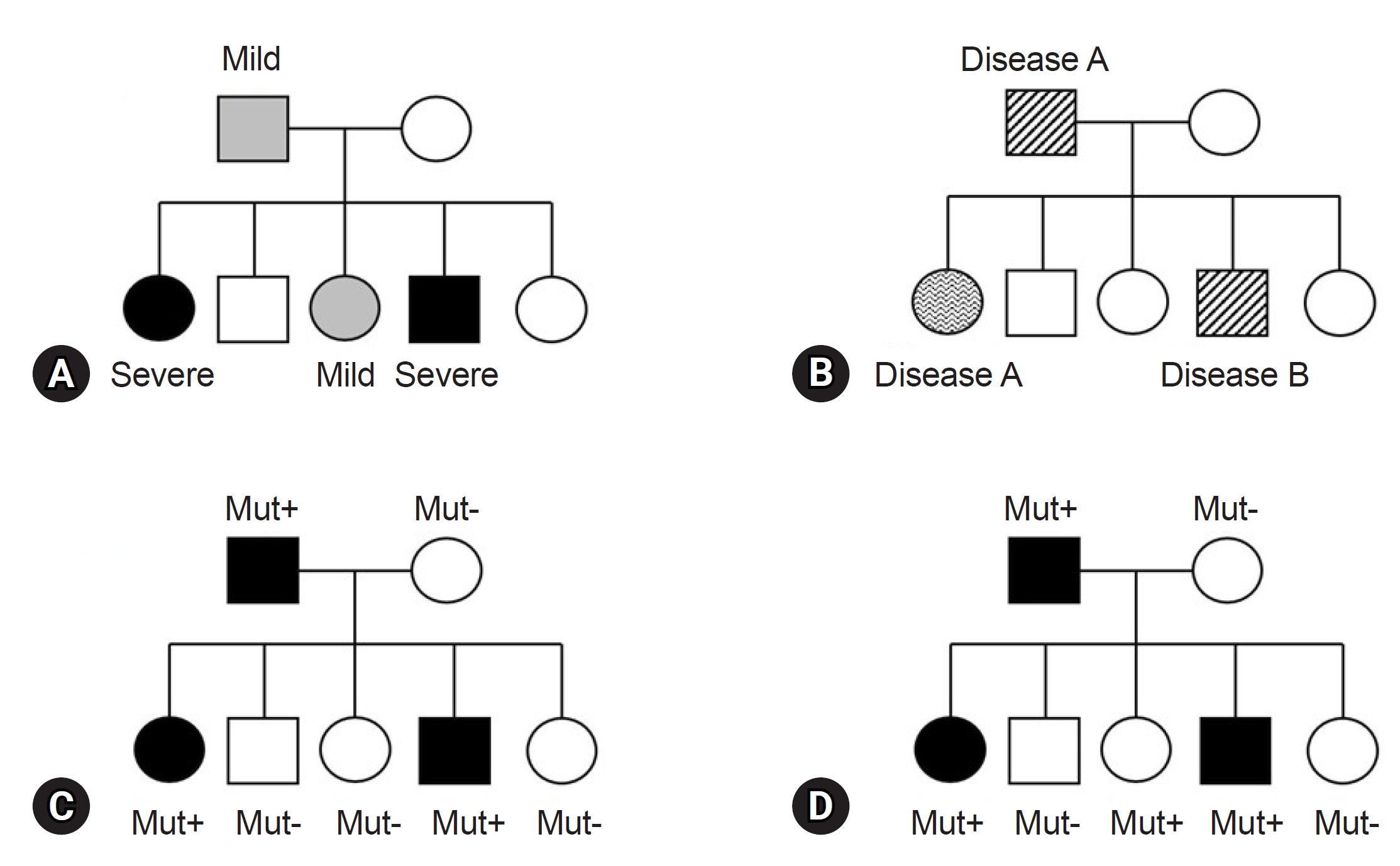
Conceptual representation of expressivity, pleiotropy, and penetrance in autosomal dominant genetic disorders. (A) A pedigree displaying an autosomal dominant genetic with varying levels of disease expressivity among family members. (B) A pedigree illustrating an autosomal dominant genetic disorder demonstrating disease pleiotropy within family members. (C) A pedigree of autosomal dominant genetic disorder exhibiting complete penetrance. (D) A pedigree of autosomal dominant genetic disorder exhibiting incomplete penetrance. Mut, mutation.
Conclusion
This review highlights the complexities and advancements in clinical genetic testing for rare diseases, emphasizing significant strides in diagnostic tool development, particularly NGS. While NGS has greatly improved our ability to identify genetic diseases, it has also necessitated meticulous validation. The selection of an appropriate genetic test in clinical practice requires careful consideration of factors such as detection range, cost, and clinical specificity. The role of germline variant classification, guided by the ACMG and ACGS guidelines, is crucial, but it faces limitations, including subjectivity and insufficient coverage of population-specific variations. To ensure accurate diagnosis and effective management, clinicians must meticulously align patient phenotypes with reported genes/variants, taking into consideration the variability in disease expression and extensive family histories (Fig. 2). This holistic and detailed approach is essential for enhancing patient care within the complex landscape of rare genetic diseases.
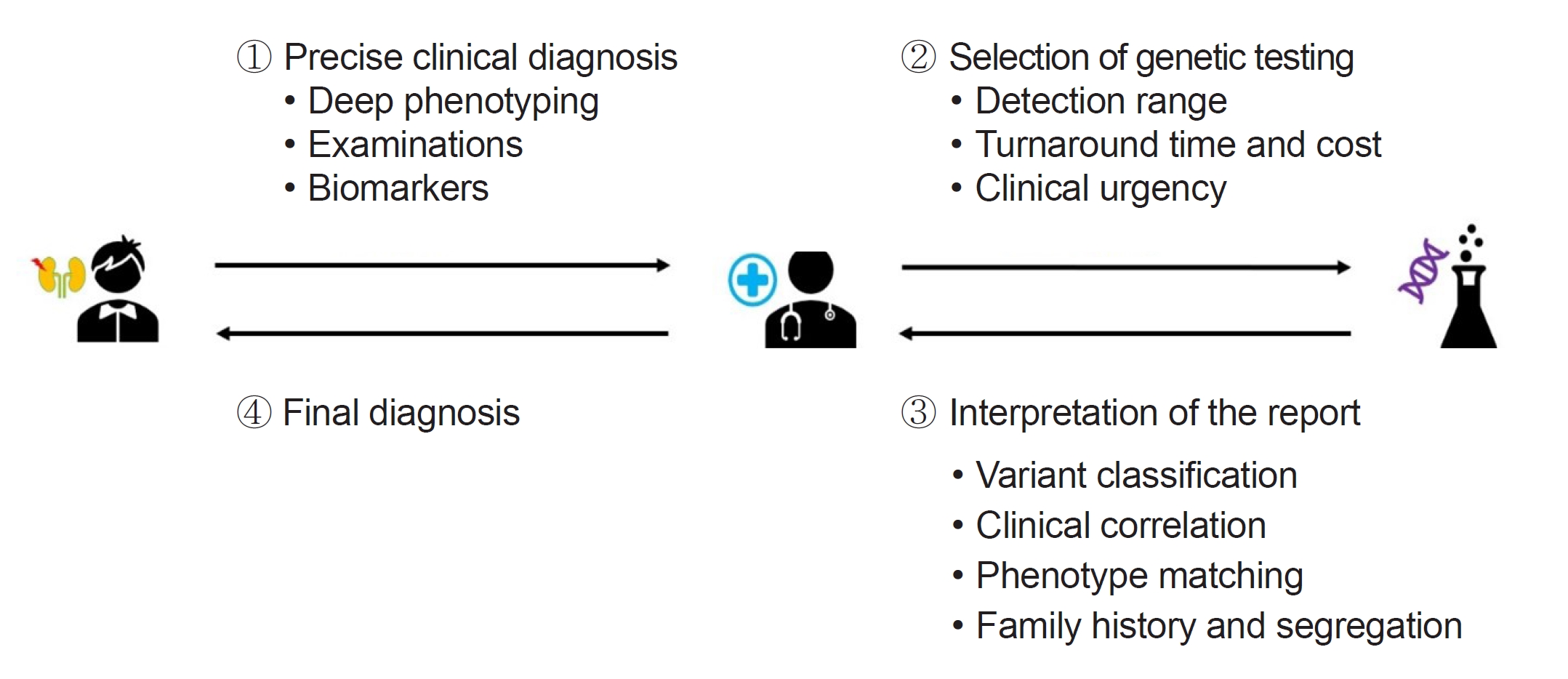
Physician’s guide to genetic testing in rare diseases.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
None.
Author contributions
All the work was done by SYK.